
Abstract
Recent studies have uncovered two signaling pathways that activate the host innate immunity against viral infection. One of the pathways utilizes members of the Toll-like receptor (TLR) family to detect viruses that enter the endosome through endocytosis. The TLR pathway induces interferon production through several signaling proteins that ultimately lead to the activation of the transcription factors NF-κB, IRF3 and IRF7. The other antiviral pathway uses the RNA helicase RIG-I as the receptor for intracellular viral double-stranded RNA. RIG-I activates NF-κB and IRFs through the recently identified adaptor protein MAVS, a CARD domain containing protein that resides in the mitochondrial membrane. MAVS is essential for antiviral innate immunity, but it also serves as a target of Hepatitis C virus (HCV), which employs a viral protease to cleave MAVS off the mitochondria, thereby allowing HCV to escape the host immune system.
Similar content being viewed by others


Introduction
Viruses are highly infectious pathogens that depend on host cellular machinery for survival and replication. Most viral infections, like the common cold caused by Rhinoviruses, are efficiently resolved by the host innate and adaptive immune systems. The innate immune response is the first line of defense against an invading pathogen. A key aspect of the antiviral innate immune response is the synthesis and secretion of type I interferons (IFN) such as IFN-α and IFN-β, which exhibit antiviral, anti-proliferative and immunomodulatory functions 1.
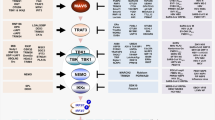
Two events required to trigger an effective anti-viral innate immune response are: a) detection of the invading virus by immune system receptors; and b) initiation of protein signaling cascades that regulate the synthesis of IFNs. The cells of the innate immune system express pattern recognition receptors (PRR) that detect invariant molecular structures shared by pathogens of various origin (pathogen-associated molecular patterns, PAMP) 2. Toll-like receptors (TLRs) 3, 7, 8 and 9 are the major PRRs that recognize distinct types of virally-derived nucleic acids and activate signaling cascades that result in the induction of type I IFNs (Figure 1) 3. Recently, retinoic acid inducible gene – I (RIG-I) has been identified as a cytosolic receptor for intracellular dsRNA 4. RIG-I induces IFN in response to intracellular viral dsRNA in a TLR-independent manner. Thus, there are two receptor systems in place to detect the presence of virus and mount an immune response. These receptor systems localize to different compartments within a cell and recognize different ligands. Our current understanding of these receptor systems in antiviral immunity and their downstream signaling cascades is the focus of this review.

TLR and RIG-I – two antiviral innate immunity pathways: Some viruses enter cells through the endocytic machinery. The genomes of such viruses are detected by members of TLR family of receptor including TLR3, TLR7, TLR8 and TLR9. These receptors have a single transmembrane domain and recognize their ligands through the leucine rich repeats (LRR) in their luminal domains. The cytoplasmic toll/IL-1 receptor (TIR) domain of these receptors enable the recruitment of adaptors such as TRIF or MyD88 that signal to downstream transcription factors ATF-2/cJun, NF-κB and IRF. RIG-I is a receptor for intracellular dsRNA. The C-terminal helicase domain of RIG-I binds dsRNA and activates the N-terminal CARD domains such that the downstream signaling cascade is initiated. MAVS is a mitochondrial protein that participates in the anti-viral signaling pathway downstream of RIG-I. Activation of either pathway leads to the induction of IFN-β.
Regulation of type I IFN gene transcription
Type I IFNs include several IFN-α subtypes and a single IFN-β subtype 1. The induction of type I IFN genes is regulated at the step of transcription and is best understood for the IFN-β promoter. A multi-protein complex called an enhanceosome is assembled at the IFN-β promoter in response to a viral challenge 5. The enhanceosome consists of at least three classes of transcription factors – ATF-2/c-Jun, nuclear factor (NF)-κB and interferon regulatory factor 3 (IRF3). Of these, the activities of NF-κB and IRF3 are regulated by their subcellular localization. In the inactive state, NF-κB is held in the cytosol by inhibitory κB (IκB) family members 6. In the presence of diverse stimuli, such as IL-1β, TNF-α, and viruses, the IκB kinase (IKK) is activated and it then phosphorylates IκB. Once phosphorylated, IκB is ubiquitinated and subsequently degraded by the proteasome. Free NF-κB then translocates into the nucleus and turns on its target genes. Similar to NF-κB, the inactive form of IRF3 is also cytosolic. In response to a viral challenge, IRF3 is phosphorylated by the IKK-like kinases TBK-1 and IKKe 7, 8. Phosphorylation of IRF3 leads to its dimerization and translocation into the nucleus. Viral infection also leads to activation of stress kinases such as JNK and p38 kinase, which phosphorylate ATF2/c-Jun in the nucleus. Together with the nuclear architectural protein HMG-I (Y), NF-κB, IRF3 and ATF2/c-Jun assemble into a stereospecific enhanceosome complex that remodels the chromatin in the promoter of IFN-β, resulting in its transcriptional initiation.
IFN-β binds to the IFNα/β receptor (IFNAR) in autocrine and paracrine manner to initiate a positive feedback loop that results in further production of type I IFNs 1. IFNARs trigger the activation of the janus kinase (JAK) family members JAK1 and Tyk-2. These kinases in turn phosphorylate and activate the signal transducer and activator of transcription 1 (STAT1) and STAT2 proteins. These transcription factors associate with IRF9 to form a heterotrimeric complex, IFN-stimulated gene factor 3 (ISGF3). ISGF3 initiates the transcription of several interferon stimulated genes (ISGs) by binding to the IFN-stimulated response elements (ISRE) in their promoter regions. The ISGs inhibit different stages of virus replication and elicit an anti-viral state in the host.
IRF7, one of the synthesized ISGs, is a member of the IRF family of transcription factors that regulates the transcription of IFN-α gene. An initial model suggested that IRF-7 is not involved in the initial phase of IFN-β induction as it is expressed at low levels in most cells in the absence of virus. IFN-β produced in response to a viral challenge by the IRF3 dependent pathway described above, induces transcription of IRF-7. IRF7 is then activated by phosphorylation at certain key residues by TBK-1/IKKe such that it binds and induces the promoter of IFN-α gene. A recent study using IRF7 knockout mice has demonstrated that transcription of both IFN-α and IFN-β is dependent on IRF7 9, indicating that IRF7 is a master regulator of type I IFNs.
IRF5 is another member of the IRF family that has been suggested to regulate type I IFN expression. However, recent genetic experiments using IRF5-deficient mice showed that IRF5 is not required for type I IFN induction, but is required for the induction of proinflammatory cytokines by stimulation of TLRs 10.
The TLR pathway of anti-viral innate immunity
The founding member of the TLRs is the Toll receptor in Drosophila, which was first found to instruct dorsal-ventral patterning in early embryos, and later found to also regulate anti-fungal innate immunity in adult flies. Sequence homology search in mammalian genomes has subsequently identified 11 members of TLRs 3. These receptors contain an extracellular domain characterized by leucine-rich repeats (LRRs), a single transmembrane domain, and an intracellular signaling domain known as the Toll/IL-1R (TIR) domain. Although all TLRs share similar extracellular LRRs, they recognize very different microbial signatures. For example, TLR3 recognizes viral double-stranded RNA, TLR4 recognizes bacterial lipopolysaccharides (LPS), whereas TLR5 is a receptor for bacterial flagellin. In most cases, however, a direct binding between a TLR and a putative microbial ligand has not been demonstrated. The intracellular TIR domain recruits signaling molecules to activate downstream signaling pathways culminating in the induction of cytokines and IFNs through NF-κB and IRFs. Except for TLR3, all TLRs utilize MyD88 as an adaptor protein to recruit downstream signaling molecules including the protein kinases IRAK4 and IRAK1, and the RING domain ubiquitin ligase TRAF6. TRAF6 functions together with a dimeric ubiquitin conjugating enzyme complex Ubc13-Uev1A to catalyze the synthesis of Lys63-linked polyubiquitin chains that lead to the activation of a protein kinase complex consisting of TAK1, TAB1 and TAB2 11, 12. The activated TAK1 kinase phosphorylates IKKβ in the activation loop, resulting in the activation of IKK and subsequent nuclear translocation of NF-κB. The TIR domains of TLR3 and TLR4 bind to another adaptor protein TRIF, which binds directly to TRAF6 and RIP1 to activate NF-κB. TRIF can also bind to TBK1, which phosphorylates and activates IRF3 and IRF7. Recent studies have also shown that TRIF and MyD88 can bind to TRAF3, which activates IRFs to induce type I IFNs, but inhibits NF-κB to suppress the induction of proinflammatory cytokines 13, 14.
Among TLRs, TLR3, 7, 8 and 9 are involved in antiviral innate immune responses 3. TLR3 recognize dsRNA viruses and may also be involved in sensing dsRNA released from dying cells. TLR7 and TLR8 are receptors for G/U-rich ssRNA associated with viruses that enter cells through endocytosis. TLR9 recognizes unmethylated CpG DNA present in DNA viruses such as herpesvirus. These receptors are localized in the endosomal membranes, with the ligand binding domain facing the lumen of the endosomes, and the TIR signaling domain positioning in the cytoplasmic side. Viral nucleic acids that arrive in this compartment through endocytosis are recognized by these receptors. The endosomal localization of TLR7, 8 and 9 is essential for signaling, as formulation of nucleic acids that allow them to retain in the endosome convert them to effective TLR ligands that induce type I IFNs 15. This explains why plasmacytoid dendritic cells (pDC) are high producers of IFNs, as these cells effectively retain viral RNA in the endosome, whereas in conventional dendritic cells (cDC), viral RNA is rapidly transported from endosome to lysosome.
The MyD88-IRAK-TRAF6 signaling module is essential for the induction of IFNs by TLR7, 8 and 9. The same signaling module is also required for the activation of NF-κB by IL-1β and other TLRs such as TLR2; however, IFNs are not induced by IL-1β or TLR2. Thus, it is likely that TLR7, 8 and 9 recruit additional components in pDCs to activate IRF7, the master regulator of IFN-α. One of such components may be TRAF3, which has recently been shown to be essential for IFN-α induction in pDCs. It has been shown that MyD88 and TRAF6 can bind to IRF7 directly, and recruit IRAK1 to phosphorylate IRF7, resulting in the nuclear translocation and activation of IRF7 16, 17. Indeed, IRAK1-deficient mice are defective in IFN-α production in response to stimulation of TLR7 and TLR9 18. Interestingly, TRAF6 appears to induce the phosphorylation of IRF7 through a mechanism that involves Ubc13-catalyzed K63 polyubiquitination 17. It would be interesting to determine if IRAK1 or another IRF7 activating kinase is activated by TRAF6 in a ubiquitin-dependent manner.
Genetic experiments clearly demonstrate that TLR7 and TLR8 are essential for interferon induction in pDCs by RNA viruses 19. However, in many other cell types, including cDCs, macrophages and fibroblasts, deletion of both MyD88 and TRIF, which abolishes all TLR signaling, has no effect on viral induction of IFNs 20. Furthermore, although human patients deficient in IRAK4 are more susceptible to bacterial infection, they have intact immune responses against viruses 21. Therefore, there must be TLR-independent pathways that are highly effective in providing antiviral innate immunity.
The RIG-I pathway of anti-viral innate immunity
Retinoic acid inducible gene I (RIG-I) has recently been identified as an intracellular receptor for viral dsRNA 4. RIG-I is a member of the DExD/H box-containing RNA helicase family of proteins that unwind dsRNA in an ATPase dependent manner. The helicase domain of RIG-I can bind both synthetic dsRNA [poly (I:C)] and viral dsRNA. Besides the C-terminal helicase domain, RIG-I also contains two tandem caspase recruitment domains (CARDs) at its N-terminus. Over-expression of the N-terminal region of RIG-I comprising the two CARD domains is sufficient to activate NF-κB and IRF3 in the absence of a viral challenge, whereas the full-length RIG-I is activated only in the presence of dsRNA. Thus, the binding to dsRNA to the RNA helicase domain of RIG-I likely induces a conformational change that exposes the N-terminal CARD domains to recruit downstream signaling proteins. Further structural analysis will be required to determine the exact mechanism of how RIG-I is regulated by dsRNA binding.
The functional significance of RIG-I in anti-viral immunity was shown first by RNAi studies and confirmed by mouse knockout studies 4, 20. RNAi of RIG-I in L929 cells, a mouse fibroblast cell line, inhibited not only IRF3 activation but also subsequent induction of type I IFNs in response to RNA viruses. The embryos of RIG-I knockout mice displayed severe liver degeneration and most were embryonic lethal. The mechanism underlying the lethal phenotype of RIG-I mutant mice is not understood as yet. In mouse embryonic fibroblasts (MEFs) and lung fibroblasts, it was shown that the induction of IFN-β and ISGs by several RNA viruses was abolished. Pre-treatment of the fibroblasts with IFN-β increased the resistance of the RIG-I deficient fibroblasts to VSV, indicating that RIG-I is required for the induction of IFN-β and does not affect the downstream IFN-β signaling pathway.
An important question is the relative importance of signaling mediated by RIG-I vs TLRs in an in vivo system. This question has been addressed by comparing the interferon induction in fibroblasts and bone marrow derived dendritic cells from RIG-I-/- vs MyD88-/- TRIF-/- mice, which lack all TLR signaling 20. The ability to induce IFN-β when challenged by NDV was severely compromised in the RIG-I-/-, but not MyD88-/- TRIF-/-, cDCs and fibroblasts. Opposite results were observed for the pDCs, which induce IFN-β normally in the absence of RIG-I, but not in the absence of MyD88 and TRIF. Thus, RIG-I and TLR pathways are not redundant, but rather mediate antiviral signaling in different cell types.
Besides RIG-I, MDA-5 and Lpg2 have also been identified as DExD/H box RNA helicases that function in the antiviral immune response 22, 23. Like RIG-I, MDA-5 also contains two N-terminal CARD domains which can activate the IFN-β promoter. The importance of MDA-5 as an anti-viral protein had been suggested based on the finding that paramyxovirus V protein binds MDA-5 and inhibits its function 24. Lpg2 lacks the CARD domain and acts as a negative regulator of the RIG-I pathway. Over-expression of Lpg2 inhibits the activation of IFN-β promoter by Sendai virus, but it does not interfere with the TLR3 signaling pathway.
MAVS signaling in the RIG-I pathway
Recent studies have identified a CARD domain containing protein that acts downstream of RIG-I. This protein, independently identified by four different groups, has been called mitochondrial anti-viral signaling protein (MAVS) 25, IFN-β promoter stimulator 1 (IPS-1) 26, virus-induced signaling adaptor (VISA) 27 and CARD adaptor inducing IFN-β (CARDIF) 28. Based on its biological function as an antiviral protein and the importance of mitochondrial localization for the function of this protein (discussed below), we will refer to this protein as MAVS in this review.
Several lines of evidence demonstrate an essential role for MAVS in the antiviral signaling pathway. First, over-expression of MAVS leads to the activation of NF-κB and IRF3, and therefore type I IFN production. Second, knockdown of MAVS expression by RNAi abolishes the induction of IFNs by viruses as well as by RIG-I over-expression. Third, the activation of kinases responsible for NF-kB and IRF3 activation is abrogated in the absence of MAVS expression. Fourth, over-expression of MAVS protects cells from the cytopathic effects of VSV, whereas RNAi of MAVS renders the cells more susceptible to killing by the virus. Further epistasis studies show that MAVS functions downstream of RIG-I and upstream of IKK and TBK1 (Figure 2).

The RIG-I – MAVS signaling pathway: RIG-I is a receptor for intracellular dsRNA. It contains a C-terminal RNA helicase domain that binds to viral dsRNA, and two tandem CARD domains at the N-terminus. The binding of dsRNA to the helicase domain presumably induces a conformational change that exposes the CARD domains to initiate a signaling cascade. MAVS is a CARD domain containing mitochondrial protein that functions downstream of RIG-I. MAVS can signal to both NF-κB and IRF3 signaling pathways by activating the IKK and TBK-1/IKKε kinase complexes. Once activated, NF-κB and IRF3 translocate into the nucleus and turn on the IFN-β gene promoter. The mechanism by which MAVS activates downstream kinase pathways is not clear, although it has been shown that the mitochondrial membrane localization of MAVS is essential for its signaling function. The importance of the mitochondrial localization of MAVS is underscored by the recent discovery that the hepatitis C virus protease NS3/4A cleaves MAVS off the mitochondria to evade the host innate immune system.
Besides the N-terminal CARD domain, MAVS also contains a proline-rich (PRO) region and a C-terminal hydrophobic transmembrane (TM) region 25. Deletion analyses have shown that the CARD domain and the TM domain are essential for the function of MAVS. Unlike the RIG-I CARD domains, overexpression of MAVS CARD domain is not sufficient to induce IFN-β. But when the CARD domain is fused with the C-terminal TM domain, this truncated 'mini-MAVS' protein, which represents just one-fourth of the total length of MAVS, is sufficient to activate the downstream pathway. The TM sequence of MAVS resembles the mitochondrial targeting sequences of several C-tail anchored mitochondrial membrane proteins, including the cell survival proteins Bcl-2 and Bcl-xL. Indeed, biochemical and microscopic imaging experiments show that the C-terminal TM domain of MAVS targets the protein to the mitochondrial outer membrane. Importantly, the mitochondrial localization of MAVS is essential for its activity because the deletion of the TM domain, which mislocalizes the protein to the cytosol, abolishes the signaling function of MAVS. When the TM domain of MAVS was replaced with the mitochondrial membrane targeting domain of Bcl-2 or Bcl-xL, the function of MAVS was fully restored, indicating that it is the mitochondrial localization but not the sequence of the TM domain that is essential for MAVS activity. This conclusion is further supported by the experiments showing that mislocalization of MAVS to other membrane compartments such as plasma membrane and endoplasmic reticulum greatly impairs the ability of MAVS to induce IFNs.
The mechanisms by which MAVS is regulated by RIG-I and how MAVS signals to downstream kinases remain to be further investigated. Although MAVS has been shown to interact with RIG-I in over-expression experiments by several groups, it has not been clearly demonstrated that endogenous MAVS and RIG-I can interact in a virus-dependent manner. The signaling mechanism of MAVS is a subject of debate at present. Two groups show that MAVS can interact with TRAF6 and both identified TRAF6 binding sites within MAVS 25, 27. However, Seth et al presented evidence that the MAVS mutant (mini-MAVS) lacking all TRAF-binding sites is still capable of inducing IFN-β. Furthermore, TRAF6-deficient cells have normal induction of IFN-β following viral infection 25, 29. Kawai et al showed that MAVS interacted with RIP-1 and FADD, and proposed that these molecules linked MAVS to IKK activation 26. However, RIP1-deficient MEF cells are also fully capable of inducing IFN-β following viral challenges 25, 30. Meylan et al reported that MAVS bound to IKKα and IKKɛ directly, but such interaction was not found by the other groups. Thus, there is no consensus mechanism that emerges from these independent studies. Further studies are clearly required to elucidate the mechanism of MAVS signaling.
The discovery of MAVS has also provided a breakthrough in the field of hepatitis C virus (HCV) research. HCV infects more than 170 million people worldwide, and approximately 80% of the infected individuals develop persistent infection 31. The persistence of HCV infection is caused in part by the suppression of the host immune system by HCV viral proteins, such as the NS3/4A serine protease. Previous studies have shown that NS3/4A inhibits the induction of IFN-β by the RIG-I signaling pathway 32, 33, 34, however, the target of NS3/4A was not known previously. Two groups have now shown that MAVS is the long-sought target of NS3/4A 28, 35. NS3/4A cleaves MAVS at Cys-508, which is located only a few residues before the mitochondrial targeting domain of MAVS. As a result of the proteolytic cleavage, MAVS is dislodged from the mitochondria and becomes an inactive cytosolic fragement. Li et al also showed that NS3/4A binds to and co-localizes with MAVS in the mitochondrial membrane, and it can cleave MAVS directly in vitro. Using a replicon cell culture system in which the RNA genome of HCV replicates autonomously, Li et al demonstrated that endogenous MAVS is indeed cleaved by NS3/4A, and that a mutation at C508 of MAVS that prevents its cleavage restores IFN induction in the replicon cells. Meylan et al also showed that transfected MAVS is cleaved in a liver cell line infected with the recently developed HCV virus. Taken together, these results show that HCV paralyzes the host immune system by cleaving MAVS off the mitochondria, further underscoring the importance of the mitochondrial localization of MAVS in its antiviral signaling.
Conclusions and perspectives
Research in the past few years has uncovered two antiviral innate immunity pathways leading to the induction of interferons. The TLR pathway operates mainly in pDCs to detect viral RNA and DNA associated with endocytosed viral particles. In most other cell types, the RIG-I pathway is essential for innate immune responses against intracellular viral replication. While the receptors for both antiviral pathways have now been identified, the signaling pathways downstream of both receptors remain to be fully elucidated. In the TLR pathway, although it is clear that MyD88, IRAK, TRAF6 and TRAF3 are essential for the induction of IFN-α in pDCs, how these proteins lead to the phosphorylation and activation of IRF7 is not understood. Similarly, in the RIG-I pathway, although MAVS is clearly an essential adaptor molecule that links RIG-I to IKK and TBK1 activation, how MAVS is regulated by RIG-I and how it activates downstream kinases remains largely unknown. While the mitochondrial localization of MAVS is essential for its signaling function, how mitochondria play a role in activating IKK and TBK1 is still a mystery. Future studies should also uncover more examples of host-virus interaction, as illustrated from the proteolytic cleavage of MAVS by the HCV protease. It is quite possible that other viruses may have also evolved to develop novel strategies to target pivotal host immune response proteins such as MAVS. Measures to counter the viral suppression of the host immune system may prove effective in the prevention and treatment of viral diseases.
References
Honda K, Yanai H, Takaoka A, Taniguchi T . Regulation of the type I IFN induction: a current view. Int Immunol 2005; 17:1367–78.
Medzhitov R, Janeway CA Jr . Decoding the patterns of self and nonself by the innate immune system. Science 2002; 296:298–300.
Akira S, Takeda K . Toll-like receptor signalling. Nat Rev Immunol 2004; 4:499–511.
Yoneyama M, Kikuchi M, Natsukawa T, et al. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat Immunol 2004; 5:730–7.
Maniatis T, Falvo JV, Kim TH, et al. Structure and function of the interferon-beta enhanceosome. Cold Spring Harb Symp Quant Biol 1998; 63:609–620.
Silverman N, Maniatis T . NF-kappaB signaling pathways in mammalian and insect innate immunity. Genes Dev 2001; 15:2321–42.
Fitzgerald KA, McWhirter SM, Faia KL, et al. IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat Immunol 2003; 4:491–6.
Sharma S, tenOever BR, Grandvaux N, et al. Triggering the interferon antiviral response through an IKK-related pathway. Science 2003; 300:1148–51.
Honda K, Yanai H, Negishi H, et al. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature 2005; 434:772–7.
Takaoka A, Yanai H, Kondo S, et al. Integral role of IRF-5 in the gene induction programme activated by Toll-like receptors. Nature 2005; 434:243–9.
Deng L, Wang C, Spencer E, et al. Activation of the IkappaB kinase complex by TRAF6 requires a dimeric ubiquitin-conjugating enzyme complex and a unique polyubiquitin chain. Cell 2000; 103:351–61.
Wang C, Deng L, Hong M, et al. TAK1 is a ubiquitin-dependent kinase of MKK and IKK. Nature 2001; 412:346–351.
Hacker H, Redecke V, Blagoev B, et al. Specificity in Toll-like receptor signalling through distinct effector functions of TRAF3 and TRAF6. Nature 2005 [Epub ahead of print].
Oganesyan G, Saha SK, Guo B, et al. Critical role of TRAF3 in the Toll-like receptor-dependent and -independent antiviral response. Nature 2005 [Epub ahead of print].
Honda K, Ohba Y, Yanai H, et al. Spatiotemporal regulation of MyD88-IRF-7 signalling for robust type-I interferon induction. Nature 2005; 434:1035–40.
Honda K, Yanai H, Mizutani T, et al. Role of a transductional-transcriptional processor complex involving MyD88 and IRF-7 in Toll-like receptor signaling. Proc Natl Acad Sci U S A 2004; 101:15416–21.
Kawai T, Sato S, Ishii KJ, et al. Interferon-alpha induction through Toll-like receptors involves a direct interaction of IRF7 with MyD88 and TRAF6. Nat Immunol 2004; 5:1061–8.
Uematsu S, Sato S, Yamamoto M, et al. Interleukin-1 receptor-associated kinase-1 plays an essential role for Toll-like receptor (TLR)7- and TLR9-mediated interferon-{alpha} induction. J Exp Med 2005; 201:915–23.
O'Neill, L. A. Immunology. After the toll rush. Science 2004; 303:1481–2.
Kato H, Sato S, Yoneyama M, et al. Cell Type-Specific Involvement of RIG-I in Antiviral Response. Immunity 2005; 23:19–28.
Yang K, Puel A, Zhang S, et al. Human TLR-7-, -8-, and -9-Mediated Induction of IFN-alpha/beta and -lambda Is IRAK-4 Dependent and Redundant for Protective Immunity to Viruses. Immunity 2005; 23:465–78.
Rothenfusser S, Goutagny N, DiPerna G, et al. The RNA helicase Lgp2 inhibits TLR-independent sensing of viral replication by retinoic acid-inducible gene-I. J Immunol 2005; 175:5260–8.
Yoneyama M, Kikuchi M, Matsumoto K, et al. Shared and unique functions of the DExD/H-box helicases RIG-I, MDA5, and LGP2 in antiviral innate immunity. J Immunol 2005; 175:2851–8.
Andrejeva J, Childs KS, Young DF, et al. The V proteins of paramyxoviruses bind the IFN-inducible RNA helicase, mda-5, and inhibit its activation of the IFN-beta promoter. Proc Natl Acad Sci U S A 2004; 101:17264–9.
Seth RB, Sun L, Ea CK, Chen ZJ . Identification and Characterization of MAVS, a Mitochondrial Antiviral Signaling Protein that Activates NF-kappaB and IRF3. Cell 2005; 122:669–82.
Kawai T, Takahashi K, Sato S, et al. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat Immunol 2005; 6:981–8.
Xu LG, Wang YY, Han KJ, et al. VISA Is an Adapter Protein Required for Virus-Triggered IFN-beta Signaling. Mol Cell 2005; 19:727–40.
Meylan E, Curran J, Hofmann K, et al. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature 2005; 437:1167–72.
Gohda J, Matsumura T, Inoue J . Cutting edge: TNFR-associated factor (TRAF) 6 is essential for MyD88-dependent pathway but not toll/IL-1 receptor domain-containing adaptor-inducing IFN-beta (TRIF)-dependent pathway in TLR signaling. J Immunol 2004; 173:2913–7.
Cusson-Hermance N, Khurana S, Lee TH, Fitzgerald KA, Kelliher MA . Rip1 mediates the Trif-dependent toll-like receptor 3- and 4-induced NF-{kappa}B activation but does not contribute to interferon regulatory factor 3 activation. J Biol Chem 2005; 280:36560–6.
Lindenbach BD, Rice CM . Unravelling hepatitis C virus replication from genome to function. Nature 2005; 436:933–8.
Breiman A, Grandvaux N, Lin R, et al. Inhibition of RIG-I-dependent signaling to the interferon pathway during hepatitis C virus expression and restoration of signaling by IKKepsilon. J Virol 2005; 79:3969–78.
Foy E, Li K, Sumpter R Jr, et al. Control of antiviral defenses through hepatitis C virus disruption of retinoic acid-inducible gene-I signaling. Proc Natl Acad Sci U S A 2005; 102:2986–91.
Foy E, Li K, Wang C, et al. Regulation of interferon regulatory factor-3 by the hepatitis C virus serine protease. Science 2003; 300:1145–8.
Li XD, Sun L, Seth RB, Pineda G, Chen ZJ . Hepatitis C virus protease NS3/4A cleaves mitochondrial antiviral signaling protein off the mitochondria to evade innate immunity. Proc Natl Acad Sci U S A 2005; 102:17717–22.
Acknowledgements
Research in the Chen laboratory is supported by grants from NIH (RO1-AI60919 and RO1-GM63692), American Cancer Society (RSG0219501TBE), and the Welch Foundation (I-1389). Zhijian J Chen is an Investigator of Howard Hughes Medical Institute and a Burroughs Wellcome Fund Investigator in Pathogenesis of Infectious Diseases.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Seth, R., Sun, L. & Chen, Z. Antiviral innate immunity pathways. Cell Res 16, 141–147 (2006). https://doi.org/10.1038/sj.cr.7310019
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.cr.7310019
Keywords
This article is cited by
-
Gene expression profile of HCT-8 cells following single or co-infections with Cryptosporidium parvum and bovine coronavirus
Scientific Reports (2023)
-
Microbiota-derived acetate enhances host antiviral response via NLRP3
Nature Communications (2023)
-
Palmitic Acid Upregulates Type I Interferon–Mediated Antiviral Response and Cholesterol Biosynthesis in Human Astrocytes
Molecular Neurobiology (2023)
-
Sex differences and similarities in the neuroimmune response to central administration of poly I:C
Journal of Neuroinflammation (2021)
-
Triterpene-Enriched Olive Extract as an Immunopotentiator in Black Sea Bream (Acanthopagrus schlegelii)
Journal of Ocean University of China (2020)
