
Abstract
Down’s syndrome (DS) presents with a constellation of cardiac, neurocognitive and growth impairments. Individuals with DS are also prone to severe infections and autoimmunity including thyroiditis, type 1 diabetes, coeliac disease and alopecia areata1,2. Here, to investigate the mechanisms underlying autoimmune susceptibility, we mapped the soluble and cellular immune landscape of individuals with DS. We found a persistent elevation of up to 22 cytokines at steady state (at levels often exceeding those in patients with acute infection) and detected basal cellular activation: chronic IL-6 signalling in CD4 T cells and a high proportion of plasmablasts and CD11c+TbethighCD21low B cells (Tbet is also known as TBX21). This subset is known to be autoimmune-prone and displayed even greater autoreactive features in DS including receptors with fewer non-reference nucleotides and higher IGHV4-34 utilization. In vitro, incubation of naive B cells in the plasma of individuals with DS or with IL-6-activated T cells resulted in increased plasmablast differentiation compared with control plasma or unstimulated T cells, respectively. Finally, we detected 365 auto-antibodies in the plasma of individuals with DS, which targeted the gastrointestinal tract, the pancreas, the thyroid, the central nervous system, and the immune system itself. Together, these data point to an autoimmunity-prone state in DS, in which a steady-state cytokinopathy, hyperactivated CD4 T cells and ongoing B cell activation all contribute to a breach in immune tolerance. Our findings also open therapeutic paths, as we demonstrate that T cell activation is resolved not only with broad immunosuppressants such as Jak inhibitors, but also with the more tailored approach of IL-6 inhibition.
Similar content being viewed by others


Main
First desc ribed by John Langdon Down in 1866, Down’s syndrome (DS) or trisomy 21 is the most common chromosomal anomaly in the USA today, affecting 1 in 700 newborn babies3,4. This extra copy of around 200 genes results in a syndrome with considerable phenotypic variability that includes intellectual disability, developmental malformations—particularly of the heart and the gut—and increased risk of Alzheimer’s disease1. As care for individuals with DS has substantially improved in recent decades5, the immune features of DS have become apparent: patients have an increased risk of severe infectious disease concomitant with a higher incidence of autoimmunity including thyroiditis (50%), coeliac disease (5%), alopecia areata (1–11%) and type 1 diabetes (1%)1,2,6.
Recent studies into the molecular mechanisms of immunological disease have focused on the overactive interferon (IFN) response7,8,9 and thymic dysfunction10 reported in individuals with DS as most IFN receptor subunits and AIRE are expressed from chromosome 21. On the innate side, monocytes from individuals with DS exhibit basal IFN-I and II signalling and hyper-respond to IFNα and IFNγ stimulation7. On the adaptive side, thymic architecture perturbations10 and T cell polarization towards differentiated subsets have been described11. Furthermore, low B cell counts in individuals with DS have been documented for decades12 and recent investigations have identified decreased proliferation and increased apoptosis in this population13.
It is now understood that a tightly regulated immune response is critical to prevent infection and excessive inflammation: the absence of a well-orchestrated immune response leads to opportunistic infections, whereas an overactive immune response leads to systemic organ damage14. Where DS lies on this spectrum of immune dysregulation and how it contributes to clinical manifestations is still largely unknown.
Down’s syndrome is a cytokinopathy
To capture the soluble immune landscape at steady-state in DS, we performed a cytokine array on plasma from individuals with DS (n = 21) and age-matched healthy control (HC, n = 10) individuals. Donors had no signs of infection at sampling. On the basis of unsupervised hierarchical clustering on 29 analytes, individuals with DS segregated into 3 distinct categories (Fig. 1a,b). One-third had widespread soluble immune dysregulation with up to 2,000-fold elevation in 22 out of the 29 markers assayed. In 9 out of 21 individuals, a subset of cytokines was significantly elevated compared with in the control individuals (including IL-13, IL-4, TNFβ (also known as LTα) IL-6 and IL-1α). Finally, the remaining five individuals with DS clustered with HC individuals. As previously reported for HC individuals15, there was a correlation between inflammatory cytokine profiles and age in DS (analysis of variance (ANOVA), P = 0.0231; Extended Data Fig. 1a). After scoring samples based on clinical immunological manifestations (Methods and Extended Data Table 1), we observed a significant association between immune scores and the cytokine-based clusters (ANOVA, P = 0.0141; Extended Data Fig. 1b). It is not yet clear whether dysregulated cytokines drive clinical immune dysfunction in individuals with DS, or vice versa.

a, Multiplex cytokine analysis using the magnetic Luminex assay of plasma from individuals with DS (n = 21) and HC individuals (n = 10), expressed as the log2-transformed fold change (FC) over the mean HC per cytokine (cyt.). Unsupervised clustering of samples and cytokines using the complete method (distance metric, Euclidean). Int., intermediate. b, PCA analysis of serum cytokines from the samples. c,d, Raw values of acute phase proteins (c) and TH2 cytokines (d) in the plasma of individuals with DS (n = 21) and HC individuals (n = 10) measured using the magnetic Luminex assay. e, The frequency of basophils expressed as the percentage of CD66b− cells (non-granulocytes) from adults with DS (n = 11) and age-matched HC individuals (n = 8). f, Raw values of IL-2 and TH1 cytokines in the plasma of individuals with DS (n = 21) and HC individuals (n = 10) measured using the magnetic Luminex assay. g,h, Multiplex cytokine analysis using the magnetic Luminex assay of plasma from blood drawn at separate timepoints from individuals with DS (n = 6) expressed as log2-transformed fold change over the mean HC individuals per cytokine followed by unsupervised clustering (g) and as raw values (h). No significant differences were detected on the basis of paired t-tests between blood draws for each individual for each cytokine (P > 0.05 for all pairs). For c–f, data are mean ± s.d. Significance was assessed using two-tailed unpaired t-tests (c–e) and ANOVA with Tukey’s post hoc analysis (f); *P ≤ 0.05; **P ≤ 0.005; ***P ≤ 0.0005; ****P ≤ 0.0001.
To determine the magnitude of this global cytokine dysregulation, we compared the cytokine profiles of individuals with DS and HC individuals at steady state, in mild or severe COVID-19 (n = 13 (control) and n = 7 (individuals with DS)) or another acute respiratory infection (n = 1 individual with DS) (Extended Data Fig. 1c). The COVID-19 samples were collected during hospitalization or at follow-up. Samples from donors with DS and with COVID-19 were available at only one point, whereas control samples were collected during the acute phase and at follow-up. Notably, the cytokine profiles of the patients with an infection (irrespective of ploidy, disease severity or infecting virus) were not significantly different from those of uninfected individuals with DS—unbiased hierarchical clustering placed patients with an infection across all three groups of individuals with DS. The uninfected high-cytokine DS group had a broader, more severe inflammatory profile compared with any patient with an infection. Our findings suggest that at least a third of individuals with DS have cytokine levels similar to severe acute infection at the baseline.
The acute-phase proteins IL-6, IL-1α and TNFβ were basally elevated in most individuals with DS (Fig. 1a,c), consistent with previous studies16,17. Intracellular staining suggested that CD16+ monocytes, conventional dendritic cells (cDCs), and central memory CD4 and CD8 T cells from individuals with DS contained slightly more IL-6 (Extended Data Fig. 1d). T helper 2 (TH2) cytokines IL-4 and IL-13, two central drivers of the allergic response18, were significantly elevated in the plasma of individuals with DS (Fig. 1a,d), possibly explained by the concurrent increase in basophils (Fig. 1e). The role of other T cell subsets, especially TH2 and TH9 cells, must be further examined. IL-2, TH1 cytokines IL-12 and IFNγ, and the TH17 cytokine IL-17 were elevated in only the high-cytokine subgroup of individuals with DS (Fig. 1a,f).
Measuring cytokine levels in multiple blood samples, drawn 5 to 10 months apart, we found that the immune profile of individuals with DS was highly stable for both the high- and low-cytokine groups (Fig. 1g,h). Our findings suggest that individuals with DS have stable, long-lasting perturbations in their cytokine levels similar to those in acute COVID-19. We conclude that DS can be considered to be a cytokinopathy.
Basal IL-6-mediated T cell activation
Cytometry by time-of-flight (CyTOF)-based immunophenotyping of whole blood from individuals with DS and age-matched HC individuals (Extended Data Fig. 2a–i) revealed that both CD4 and CD8 T cells in DS were skewed towards a memory phenotype (Fig. 2a,b and Extended Data Fig. 2d,e). There were fewer naive CD4 and CD8 T cells in individuals with DS, with a concurrent increase in CD4 central memory frequency11,19. We also uncovered baseline phosphorylated STAT3 (pSTAT3) in naive, activated and central memory CD4 T cells of individuals with DS, suggesting active cytokine signalling (Fig. 2c and Extended Data Fig. 3a).

a,b, Representative plots and calculated frequencies of CD4+ (a) and CD8+ T cell naive, central memory (CM), effector memory (EM) and terminally differentiated effector memory (TEMRA) (b) subsets in whole blood from adults with DS (n = 10) and age-matched HC individuals (n = 8). c, Basal STAT3 phosphorylation in CD4+ T cell subsets from individuals with DS (n = 19) and age-matched HC individuals (n = 13) and expressed as the log2-transformed fold change over the mean of HC individuals per subset. d–g, STAT3 phosphorylation in CD4+ T cell subsets from individuals with DS, normalized to the maximum value per experiment after ex vivo whole-blood treatment for 4 h with JAK inhibition (tofacitinib) (500 nM) (n = 7) (d); IFN blockade (anti-IFNAR2 (5 μg ml−1), anti-IFNα (0.2 μg ml−1) and anti-IFNβ (0.2 μg ml−1) antibodies) (n = 6) (e); IL-10 blockade (anti-IL-10 (5 μg ml−1) and anti-IL-10R (5 μg ml−1) antibodies) (n = 6) (f) or IL-6 blockade (tocilizumab, 50 μg ml−1) (n = 7) (g). In a and b, the whiskers denote the minimum and maximum values, the box limits denote quartiles 1–3, and the centre bar denotes the mean. For a–g, significance was assessed using two-tailed unpaired t-tests; NS, not significant (P > 0.05).
STAT3 is a transcription factor that is activated downstream of multiple cytokines and growth factors20. When aberrantly phosphorylated, STAT3 contributes to lymphoproliferation, recurrent infections and increased autoimmunity including eczema, type 1 diabetes and hypothyroidism21. Clinically, Mendelian STAT3 gain of function (GOF) substantially overlaps with DS, indicating that engagement of STAT3 in CD4 T cells in DS may contribute to immune pathogenesis. The molecular mechanism of disease in STAT3 GOF is still debated, but STAT3 attenuation of STAT5 phosphorylation is the leading hypothesis. In DS, basal phosphorylated STAT5 (pSTAT5) levels were unaffected in CD4 T cells and even increased in CD4 TEMRAs (Extended Data Fig. 3b), suggesting that other factors are involved.
To test whether basal pSTAT3 is caused by cytokine signalling instead of an intrinsic STAT3 GOF in DS, we incubated whole blood with the FDA-approved Jak inhibitor tofacitinib. This treatment restored basal pSTAT3 to control levels, indicating that STAT3 activation in DS is Jak dependent (Fig. 2d and Extended Data Fig. 3c).
Given the detection of IFNα2 and IL-10 in a subset of individuals with DS (Fig. 1a), combined with the triplication of their cognate receptors (IFNAR1, IFNAR2 and IL10RB) in DS, we hypothesized that basal pSTAT3 was due to heightened IFN-I or IL-10 response. However, IFN-I blockade did not affect basal pSTAT3 (Fig. 2e). Blocking IL-10 resulted in a modest decrease in pSTAT3 that was most pronounced in effector memory CD4 cells (Fig. 2f), suggesting that IL-10 may contribute to baseline signalling in DS. These blocking experiments were performed ex vivo, so the effect of these cytokines over time may not have been captured.
Having established that basal pSTAT3 is initiated upstream of Jaks and largely independent of IFN-I and IL-10, we turned to the IL-6–STAT3 axis. IL-6 is a potent inducer of pSTAT3 and was significantly elevated in DS (Fig. 1a,c). IL-6 blockade with the FDA-approved IL-6 receptor (IL-6R) inhibitor tocilizumab fully abrogated pSTAT3 (Fig. 2g and Extended Data Fig. 3d), indicating that IL-6 is a major mediator of baseline CD4 activation in DS. Furthermore, there was no hyper-response to exogenous IL-6 stimulation and no increase in IL-6R expression in DS (Extended Data Fig. 3e,f), confirming that elevated pSTAT3 is a result of high IL-6 rather than an intrinsic gain of expression or function in IL-6R. Together, these data implicate IL-6 signalling in ongoing CD4 T cell activation in DS and offer a more specific therapeutic target than Jak inhibition for consideration by physicians treating individuals with DS.
Notably, genetic alterations of STAT3 signalling also lead to a disturbance of B cell subsets, with a decrease in a recently described atypical B cell activation in adults with STAT3 loss of function and a corresponding increase in these CD11c+ B cells in STAT3 GOF. Given our findings of basal pSTAT3 in DS T cells, the overlap in clinical manifestations between DS and STAT3 GOF, and previously documented B cell disturbances in DS13, we further investigated the B cell compartment.
Increased CD11c+TbethighCD21low B cells
Total B cells were profoundly decreased in individuals with DS (Fig. 3a,b and Extended Data Fig. 4a) as seen previously22,23. All whole-blood samples were fixed before freezing, which enabled us to analyse all B cell populations including plasmablasts. The relative frequencies of B cell subsets were altered in DS: memory B cells were slightly decreased compared with HC individuals, whereas plasmablasts increased by almost threefold (Fig. 3a,c). Given the unexpected co-occurrence of elevated plasmablasts and depleted memory cells, combined with the simultaneous widespread elevation of pro-inflammatory cytokines and overactivation of CD4 T cells discussed above, we examined the possibility that B cells undergo inordinate activation in DS.

a, Representative t-distributed stochastic neighbour embedding (t-SNE) analysis of a fixed number of B cells to illustrate subset distribution in whole blood from adults with DS and age-matched HC individuals. b,c, The frequency in adults with DS (n = 10) and age-matched HC individuals (n = 10) of total B cells expressed as the percentage of CD66b− cells (non-granulocytes) (b) and B cell subsets expressed as the percentage of total B cells (c). d–g, The frequency in HC adults (n = 10), adults with DS (n = 10) and patients with SLE (n = 6) of CD11c+ B cells in the IgD+ naive (CD27−CD38lowIgD+) or DN (CD27−CD38lowIgD−) compartments expressed as the percentage of total (d), IgD+ naive (e) or DN (f) B cells, and the log2-transformed ratios of CD11c+ subsets to CD11c− subset (rN:aN and DN2:DN1) (g). h–k, Intracellular Tbet expression (h), and surface expression of FAS and CD86 (i), CD21 (j) and CXCR5, CCR7, CCR4 and CXCR3 (k) in naive and DN B cells from both HC individuals (n = 2–4) and individuals with DS (n = 3–10). MSI, mean signal intensity; MFI, mean fluorescence intensity. l,m, Correlation of CD11c+ B cells and circulating IL-6 (l) and cTFH1/17 (m). r, Pearson correlation coefficient. In b–g, the whiskers denote the minimum and maximum values, the box limits denote quartile 1 to quartile 3, and the centre bar denotes the mean. Significance was assessed using two-tailed unpaired t-tests (b and h–k) and one-way ANOVA with Tukey’s post hoc analysis (d–g).
CD11c+ B cells are thought to derive from naive B cells by cytokine and T cell stimulation and/or TLR engagement outside of germinal centres24. They are also characterized by high Tbet and low CD21 expression and can differentiate into plasmablasts25. The two major documented subsets of CD11c+ B cells are CD27−IgD+ activated naive (aN) B cells and a subpopulation of class-switched CD27−IgD− (double-negative (DN2)) B cells26,27 (Extended Data Fig. 4b). This B cell activation is a hallmark of the heterogenous condition systemic lupus erythematosus (SLE). It has also been described in other autoimmune and autoinflammatory diseases including rheumatoid arthritis, ulcerative colitis and common variable immunodeficiency25,27,28.
Our analysis revealed increased frequencies of CD11c+ B cells in both the IgD+ naive and double-negative (CD27−IgD−) compartments in individuals with DS compared with in HC individuals (Fig. 3d–f), at a frequency comparable to mild SLE (Fig. 3d–f). Elevation of previously described CD11c+ intermediates (aN and DN2 B cells) was concurrent with a reduction in traditional B cell activation, evidenced by decreased frequencies in resting naive (rN) and DN1 B cells. The rN:aN and DN1:DN2 ratios were significantly lower in individuals with DS and SLE compared with in HC individuals (Fig. 3g). Despite the inherently variable cell frequencies in childhood, aN B cells were significantly higher in children with DS, and DN2 B cells trended upward (Extended Data Fig. 4c–g), demonstrating that this atypical B cell activation also occurs early in life.
We next performed high-dimensional analysis of functional markers of CD11c+ B cells. As reported previously, both aN and DN2 B cells displayed upregulation of the IFNγ-induced transcription factor Tbet (Fig. 3h), and the surface receptors FAS (also known as CD95) and CD86 (also known as B7-2)29 (Fig. 3i). These cells also exhibited lower expression of CD21 (Fig. 3j). Expression of chemokine receptors in CD11c+ cells was consistent with previously described extrafollicular activation. The follicle-homing receptors CXCR5 and CCR7 were significantly downregulated in aN and DN2 B cells compared with in rN and DN1 B cells (Fig. 3k), concurrently with an increase in CXCR3 in aN cells (Fig. 3k), which suggests homing potential to inflamed tissues30. We also found elevated expression of a marker in aN B cells, CCR4 (Fig. 3k), a receptor that is classically associated with CD4 TH2 cells and is thought to be involved in homing to the skin and lungs31,32. Further research is needed to elucidate whether CCR4 expression in these B cells can drive pathology.
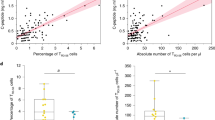
Although CD11c+ B cell frequency in DS was not correlated with age27,33 (Extended Data Fig. 4h), it was correlated with total cytokine levels (Extended Data Fig. 4i), specifically IL-6 levels (Fig. 3l), as well with the frequency of circulating T follicular helper 1/17 (cTFH1/17) cells (Fig. 3m), as in other primary immunodeficiencies28. This suggests that cytokines and activated CD4 T cells contribute to putative extrafollicular B cell differentiation24. Finally, CD11c+ B cell frequency was correlated with the number of autoimmune manifestations in our cohort (Extended Data Fig. 4j), indicating their potential role in autoimmunity.
In conclusion, CD11c+ B cells are a prominent component of the abnormal B cell response in DS. On the basis of our research and previous work in other inflammatory conditions25,28, we hypothesize that this dysregulated B cell response weakens immune tolerance and, ultimately, contributes to autoimmunity in DS.
Ex vivo naive B cell differentiation
CD11c+ B cells can differentiate into antibody-secreting plasmablasts with little to no affinity maturation, potentially explaining their association with autoimmunity34. In vitro, we confirmed that stimulated CD11c+ B cells sorted from HC peripheral blood mononuclear cells (PBMCs) differentiated into plasmablasts and secreted immunoglobulin G (IgG), in contrast to treated naive cells (Fig. 4a,b). This was potentiated by the addition of IFNγ (Fig. 4a), as previously published27. In whole blood, CD11c+ B cell frequency was positively correlated with that of plasmablasts (Fig. 4c). We also confirmed that total IgG is elevated in DS plasma compared to HC plasma13 (Extended Data Fig. 4k).

a,b, Plasma cell differentiation (a) and secreted IgG in the supernatant (b) after culture for 4 days of sorted HC naive or CD11c+ B cells in the presence of BAFF, IL-2, IL-10, IL-21, the TLR7/8 ligand R848 with or without IFNγ. n = 3 biologically independent samples. Norm., normalized. c, Correlation of CD11c+ B cell and plasmablast frequencies in adults DS (n = 12) and age-matched HC individuals (n = 8). d, Plasma cell differentiation after culture for 6 days of sorted HC naive B cells in the presence of BAFF, IL-2, IL-10, IL-21, R848 and IgG-depleted plasma from HC individuals (n = 2) or individuals with DS (n = 4). e, Magnetic-activated cell sorting (MACS)-isolated naive B cells from a healthy donor were cultured for 3 days with BAFF, IL-2, IL-21, R848, anti-IgM and plasma of HC individuals or individuals with DS in the presence of a combination of antibodies blocking IFN-I, IFN-II, IL-6 and TFNα signalling, in triplicates. Significance was assessed using two-tailed unpaired t-tests. Data are mean ± s.e.m. The results are representative of two independent experiments with n = 2 donors per group. f–h, Co-cultures containing T cells activated with IL-6, IL-2, both or polarized into TH1 cells with IL-2, IL-12 and anti-IL-4 together with MACS-isolated naive B cells from the same donor, run in triplicates. The frequency of plasmablasts (f) and extracellular CD11c induction (g) and downregulation of CD21 expression (h) in non-plasmablast B cells after co-culture for 3–6 days. Significance was assessed using one-way ANOVA with Tukey’s post hoc analysis. i,j, CD11c induction (i) and plasmablast differentiation (j) in naive CD11c− B cells isolated from controls (n = 4) or individuals with DS (n = 2) after 3 days of co-culture with CD4 T cells isolated from the same donors (syngeneic cultures). For b, d and f–j, data are mean ± s.d.
We next performed experiments to test the mechanisms underlying increased B cell activation in DS. To first address whether the cytokines in DS plasma could induce this differentiation, we co-incubated naive or total B cells sorted from control PBMCs in extrafollicular-stimulating conditions27 in the presence of IgG-depleted plasma derived from HC individuals or individuals with DS. There was significantly more plasmablast differentiation in the presence of DS plasma compared to HC plasma (Fig. 4d and Extended Data Fig. 5a). Exogenous cytokine treatment could also influence plasmablast differentiation: IFNα and IFNγ potentiated differentiation, whereas IL-4 restricted it35,36 (Extended Data Fig. 5b–d). Individual blocking of IL-6, IFN-I, IFNγ and TNF in donor plasma had no effect, whereas Jak inhibitors inhibited plasmablast differentiation, suggesting that a few cytokines are involved (Extended Data Fig. 5f). Blocking a combination of four cytokines (IL-6, IFN-I, IFNγ and TNF) reversed the DS plasma-induced augmentation of plasmablast differentiation (Fig. 4e). Thus, the cytokines present in DS plasma—at least, IL-6, IFN-I, IFNγ and TNF—are drivers of naive B cell differentiation into plasmablasts.
Given the mounting evidence in vivo that T cells drive extrafollicular B cell activation24,28, together with our findings of basal IL-6 signalling in DS CD4 T cells, we tested whether T cells contribute to this B cell response. Previous studies have demonstrated that TH1-polarized T cells can induce extrafollicular differentiation of naive B cells in vitro33,37. When we modelled DS CD4 T cell activation with exogenous IL-6, co-culture of naive B cells and T cells pretreated with IL-2 and IL-6 resulted in plasmablast differentiation equivalent to that of TH1–B cell co-cultures (Fig. 4f). Exogenous IL-6 alone did not affect plasmablast differentiation (Extended Data Fig. 5b). Furthermore, TH1-cell- and IL-6-primed CD4 T cells induced CD11c expression in co-cultured B cells (Fig. 4g), together with CD21 downregulation (Fig. 4h) and a slight increase in Tbet (Extended Data Fig. 5g). Although IFNγ is thought to have a major role in the TH1-mediated atypical B cell differentiation and was indeed present in the TH1 supernatants, we did not detect IFNγ in the IL-6 conditions (Extended Data Fig. 5h), which indicates that other cytokines drive B cell activation in these conditions. Together, these results demonstrate that cytokines and T cells in combination can drive an extrafollicular B cell response.
To better replicate the physiological conditions of DS, we performed these experiments with pre-incubation of T cells in plasma derived from individuals with DS. We found that these cells induced increased plasmablast differentiation of naive B cells compared with T cells incubated in control plasma (Extended Data Fig. 5i). Finally, to determine whether activated CD4 T cells of individuals with DS are poised to induce differentiation of naive B cells, we isolated CD4 T cells from control individuals (n = 4) and individuals with DS (n = 2) and co-cultured them with CD11c− naive B cells in syngeneic co-cultures. Without exogenous polarization, T cells from individuals with DS induced more CD11c expression and plasmablast differentiation than those of HC individuals (Fig. 4i,j). These data indicate that cytokine milieu and steady-state cellular activation contribute to atypical B cell activation in DS.
Autoimmune features in CD11c+ B cells
Next, we looked at immunoglobulin isotype expression to further ascertain the antibody-secreting potential and the naivety of CD11c+ B cells. The frequency of IgD+CD11c+ B cells was intermediate between naive and memory B cells (Fig. 5a). The proportion of IgA+CD11c+ B cells was similar to memory B cells (Fig. 5b), demonstrating that a portion of these atypical cells have gone through class switching and are therefore probably antigen experienced. We did not detect significant differences in isotype usage between HC and DS CD11c+ B cells (Extended Data Fig. 6a).

a,b, Expression of IgD (a) and IgA (b) in naive, CD11c+ and memory B cells and plasmablasts from adults with DS and age-matched HC individuals. n = 3 each. c–h, BCR sequencing analysis of genomic DNA isolated from sorted naive, CD11c+ and memory B cells from controls (n = 6) and individuals with DS (n = 6). c, The fraction of productive BCRs represented more than once in each sample. Individuals from whom a sample had fewer than 1,000 productive templates were excluded. d, CDR3 length of productive BCRs in B cells subsets in HC and DS, expressed as the number of nucleotides (nt). e, The mean number of nucleotides different from reference in the V gene of productive BCRs in B cells subsets in HC and DS. f, The frequency of productive BCRs that were aligned to the IGHV4-34 gene in each sample. g, 9G4 surface expression in naive, CD11c+ and memory B cells from HC individuals (n = 3) and individuals with DS (n = 3). h, ELISA quantification of 9G4 antibodies in the plasma of HC individuals (n = 8), and individuals with DS in the low/medium (n = 7) and high (n = 5) cytokine groups, expressed as the fold change over HC individuals. OD490, optical density at 490 nm. For a, b, g and h, data are mean ± s.d. For d–f, the whiskers denote the minimum and maximum values, the box limits denote quartile 1 to quartile 3, and the centre bar denotes the mean. Significance in a and h and significance between cell subsets in d was assessed using one-way ANOVA with Tukey’s post-hoc analysis. Significance in d–g between the HC and DS groups was assessed using two-tailed paired t-tests.
To assess the clonality of CD11c+ B cells, we performed B cell receptor (BCR) sequencing (BCR-seq) of DNA from sorted naive, CD11c+ and memory B cells from individuals with DS (n = 6) and age-matched control individuals (n = 6) at steady state (Extended Data Fig. 6b,c). In naive cells, around 99% of BCRs were unique, whereas, in CD11c+ cells and memory cells, up to 7% of BCRs were expanded (Fig. 5c), suggesting that, like memory B cells, CD11c+ B cells undergo clonal expansion (or are the result of clonal expansion) rather than non-specific stimulation. There was no difference in clonality in CD11c+ B cells between HC individuals and individuals with DS, as evidenced by similar fractions of non-unique BCR templates (Fig. 5c) and similar Simpson diversity index metrics (Extended Data Fig. 6d). The complementarity determining region 3 (CDR3) repertoires of CD11c+ and memory B cell subsets overlapped at the nucleotide and amino acid levels (Extended Data Fig. 6e,f), demonstrating that cells in these two phenotypically defined subsets derive from the same lineage. In future studies, BCR-seq analysis of donors immunized with a known antigen will enable closer examination of clonal expansions of these B cells.
The median CDR3 length of CD11c+ B cells was similar to that of naive cells and significantly longer than that of memory cells (Fig. 5d). It was not significantly different between these cells in individuals with DS and HC individuals. Increased CDR3 length in antibody-secreting cells is associated with antibody polyreactivity and autoimmunity38, suggesting that these CD11c+ B cells are prone to autoreactivity.
The number of non-reference nucleotides in the V genes of CD11c+ B cells was intermediate between that of naive and memory B cells (Fig. 5e). Notably, CD11c+ B cells in individuals with DS had significantly fewer non-reference nucleotides compared with in HC individuals. This may indicate a comparative lack of somatic hypermutation, which could lead to a broader, more non-specific humoral response. Our method was limited to CDR3 sequencing; thus, characterization of the full antigen-binding region is still needed to ascertain the true somatic hypermutation rate.
Analysis of BCR V gene usage (Extended Data Fig. 6g) revealed that CD11c+ B cell expansion is probably more autoreactive in individuals with DS than in HC individuals. There was a significantly higher usage of the IGHV4-34 gene, which is associated with autoreactivity27,38, in CD11c+ B cells in DS (Fig. 5f). We confirmed these BCR-seq results using flow cytometry: 9G4 idiotype antibodies encoded by this IGHV4-34 gene segment were more highly expressed in these atypical B cells in DS (Fig. 5g). In vitro stimulation of sorted CD11c+ B cells into antibody-secreting cells led to higher 9G4 secretion than from memory B cells (Extended Data Fig. 4h), further accentuating the link between these rare B cells and autoimmune potential. Circulating 9G4 antibodies were also elevated in the plasma of individuals with DS and were more abundant in the high-cytokine individuals with DS than in the low/intermediate groups (Fig. 5h). 9G4 antibodies are known contributors of autoimmunity in SLE, displaying specificity for nuclear antigens, dsDNA and apoptotic cells39,40. In conclusion, CD11c+ B cells in DS are present at a higher frequency and are more likely to exhibit features of self-reactive BCRs.
Autoantibodies are enriched in DS
Given clinical autoimmunity in DS and our findings of cytokine dysregulation and autoimmune-prone B cells, we hypothesized that trisomy 21 results in the generation of autoantibodies. To characterize the DS autoreactive repertoire, we assessed the plasma IgG and IgA reactivity of individuals with DS (n = 5), age-matched healthy individuals (n = 4), 3 individuals with immunodysregulation polyendocrinopathy enteropathy X-linked syndrome (IPEX) and 1 individual with autoimmune polyglandular syndrome type 1 (APS-1) (disease controls; n = 4) against >21,000 conformationally intact human proteins (CDI HuProt protein microarray) (Fig. 6a and Extended Data Fig. 7a–d).

a, PCA analysis of the HuProt IgG dataset for adults with DS (n = 5), age-matched HC individuals (n = 4), and patients with IPEX (n = 3) and APS-1 (n = 1). b, The number of IgG autoantigens enriched at least twofold in individuals with DS, IPEX and APS-1 compared with HC individuals. c, Enriched IgG autoantigens overlapping between disease groups. d, Enriched IgG autoantigens in HC individuals and in individuals with DS, IPEX and APS-1. The colour intensity corresponds to the log2-transformed fold change expression value relative to the mean of healthy adult controls. F, female; M, male; NA, unknown. e, Chromosomal expression pattern of IgG autoantigens enriched in DS. f, Gene expression pattern of IgG autoantigens enriched in DS (n = 5) according to the Human Protein Atlas. g, GO analysis of IgG autoantigens enriched in DS ranked by the number of autoantigens found to be enriched in the associated gene set. The dot size and colour intensity correspond to the FDR-adjusted P value. Med., mediated; NK, natural killer; reg., regulation; sig, signalling; surf, surface; sys., system. h, The surface expression of CD64 in monocytes, natural killer cells from individuals with DS (n = 14) and age-matched HC individuals (n = 8). Mono, monocytes. i, ELISA analysis of anti-IFNGR2 autoantibodies in the plasma from adults with DS (n = 4) and age-matched HC individuals (n = 7). j, Neutralizing IFNγ signalling in THP-1 cells by IgG fraction of plasma from adults with DS (n = 3), age-matched HC individuals (n = 3) or recombinant anti-IFNGR2 antibody. Significance was assessed using one-way ANOVA with Tukey’s post hoc analysis. For h and i, significance was assessed using two-tailed unpaired t-tests. For h and j, data are mean ± s.d. For i, the whiskers denote the minimum and maximum values, the box limits denote quartile 1 to quartile 3, and the centre bar denotes the mean.
Principal component analysis (PCA) grouped DS samples together and away from HC individuals (Fig. 6a), indicating differential self-antigen binding. The DS samples clustered with samples from patients with IPEX (n = 3), and away from the sample from the individual with APS-1 (n = 1) (caused by mutation in AIRE), although the low sample number is an important caveat (Fig. 6a).
Analysis of differentially abundant autoantigens in individuals with DS compared with HC individuals (log2-transformed fold change > 1, P < 0.05) yielded 365 proteins, in contrast to 257 and 829 proteins for APS-1 and IPEX, respectively (Fig. 6b). This indicates that autoimmunity in trisomy 21 is on par with bona fide autoimmune diseases. We observed little overlap of autoantigens between DS and APS-1 (2 out of 257), whereas a large proportion of the DS autoantigens overlapped with those in IPEX (228 out of 829) (Fig. 6c,d), a syndrome of severe multiorgan inflammation caused by defects in T regulatory (Treg) cells. The highly similar autoantibody repertoires points to T cell overactivation as a possible driver of autoreactive B cells in DS. Consistent with this finding, absolute Treg cell counts were decreased in individuals with DS compared with in HC individuals, despite similar Treg cell frequency41 (Extended Data Fig. 2f,g). Finally, 120 autoantigens were unique to DS, including metabolic (TPH1, ATP5F1, PLPP2), cell signalling (RRAGC, GPR143, PTGER4), immune signalling (DTX4, CCL11, TROVE2) and neuronal pathway intermediates (ABAT, ATP6V1G2, KCTD7) (Fig. 6c,d and Supplementary Data 1 and 2).
The autoantigens enriched in DS were encoded across all 23 chromosomes (Fig. 6e). They included proteins expressed at sites of clinical autoimmunity in DS, such as the gastrointestinal tract (MUC4, ENPP3, FABP6) and the pancreas (AMY1A, CELA3A, CTRL) (Fig. 6f). The four individuals with DS with hypothyroidism assayed did not have known anti-thyroid autoantibodies in contrast to previously described individuals with DS42 but, instead, had antibodies directed against proteins non-specifically expressed in the thyroid (CALR, LYSMD4, CYP20A1) (Fig. 6f). We also saw an enrichment of autoantigens expressed in the central nervous system (CNS) (ATP1B2, LRRC4C, ATP6V1G2) (Fig. 6f). Although antibodies cannot cross the intact blood–brain barrier, pathology can be mediated through a weakened blood–brain barrier43. Erroneous expression of normally neuronally restricted markers through altered gene expression in DS is another potential mechanism44. Given the prominent central nervous system features of DS, further investigation is warranted. Finally, immune autoantigens were prominently enriched in DS, including IFNGR2, TLR9, CD1C and IL-1R2 (Fig. 6f,g and Extended Data Fig. 7e). Immune system process, cell-surface receptor signalling and immune response were the most significantly enriched Gene Ontology (GO) terms (Fig. 6g). Abundant autoantibodies directed against the immune system may promote further immune dysregulation.
On the cellular side, we detected increased expression of high-affinity FCGR1A (also known as CD64) in monocytes and natural killer cells in DS (Fig. 6h). This receptor can engage bound antibodies and immune complexes to trigger potent inflammation and tissue injury45. Cells in individuals with DS may be poised to cause tissue damage based on the observed expression patterns.
We next validated the HuProt array results using an enzyme-linked immunosorbent assay (ELISA), confirming the presence of anti-IFNGR2, MSTN and ATP6V1G2 autoantibodies in individuals with DS but not in HC individuals (Fig. 6i and Extended Data Fig. 7f). We then functionally tested anti-IFNGR2 autoantibodies (enriched 2.7-fold in DS, false-discovery rate (FDR) = 0.01): compared to HC IgG, DS IgG resulted in a significant decrease in the response to IFNγ as measured by pSTAT1 (Fig. 6j). This demonstrates that immune-targeting autoantibodies in individuals with DS can directly inhibit the activity of cytokines. As genetic defects in IFNγ signalling cause susceptibility to mycobacterial disease46, anti-IFNGR2 autoantibodies may predispose individuals with DS to mycobacterial disease, which should be investigated in DS especially as individuals age.
Given the increased susceptibility to severe COVID-19 in individuals with DS47, we examined anti-IFN-I autoantibodies. These were modestly enriched in individuals with DS compared with HC individuals in the CDI array, but much less so than in APS-1 (Extended Data Fig. 7g). We confirmed anti-IFNα2 and anti-IFNω autoantibody enrichment in DS by Gyros assay, at titres over tenfold lower than that of APS-1 samples (Extended Data Fig. 7h). Plasma from individuals with DS with or without COVID-19 was insufficient to inhibit IFN-I, whereas samples from the individual with APS-1 fully abrogated IFN-I signalling. We noted only mild neutralization of very low amounts of IFNα2 (100 pg ml−1) by DS plasma (Extended Data Fig. 7i).
In conclusion, we detected a prominent autoantibody landscape in the plasma of individuals with DS. Together with our findings of B and T cell activation, we hypothesize that this broad humoral response predisposes individuals with DS to form high-affinity, tissue-specific antibodies that manifest clinically as autoimmune disease.
Discussion
Autoantibodies in DS have long been implicated in many of the syndrome’s features, such as thyroid disease, type 1 diabetes and even cognitive decline42,48,49,50. Here we delineate the predisposition to autoreactivity and identify the autoantibody repertoire in DS (Extended Data Fig. 8). Two of the dysregulated immune features that we detected—the cytokine landscape and CD11c+ B cell frequency—had a direct moderate association with clinical immune manifestations (P = 0.0141 by ANOVA and P = 0.0795 by r2 test, respectively). Perhaps dampening chronic steady-state inflammation in DS could prevent the accumulation of self-reactive autoantibodies and lessen disease burden.
We identified an increased frequency of CD11c+Tbet+CD21low B cells as a hallmark of immune dysregulation in DS and established a link between these cells and the elevated plasmablast frequency and increased IgG in DS plasma. Others have also implicated these cells in the production of self-reactive antibodies34, which may partly explain autoimmune susceptibility in DS. Beyond the increased frequency of these intrinsically pro-inflammatory cells in DS, BCR sequencing revealed that CD11c+ cells are more likely to have self-reactive features. Thus, in DS, these putatively extrafollicular B cells are skewed towards autoimmunity in both quantity and quality. Their direct involvement in the generation of autoantibodies and their potential damage at the tissue level merit further investigation.
We uncovered activation of naive B cells not only by cytokines in DS plasma but also by CD4 T cells with basal pSTAT3. Patients with STAT3 loss-of-function mutations have fewer CD11c+ B cells, whereas those with STAT3 GOF mutations have more28, like individuals with DS. Although it is unclear whether the exaggerated naive B cell activation in STAT3 GOF is due to intrinsic STAT3-mediated signalling in B cells, stimulation by overactive T cells or both, we hypothesize that baseline T cell activation in DS has an important role in this B cell response. This notion is further emphasized by the similarity in autoantibody repertoires between DS and IPEX, an autoimmune disease mediated by lack of T cell regulation. Our in vitro experiments demonstrate that IL-6 activation of T cells is sufficient to drive the differentiation of naive B cells into CD11c+ B cells and plasmablasts. Although the physiological implications of this model must be firmly tested, these data provide a mechanistic link between IL-6 activation of T cells and this atypical B cell response. Thus, resolving basal T cell activation in DS with a Jak inhibitor or IL-6 blockade is a promising therapeutic avenue to temper hyperinflammation and the autoimmune feed-forward loop that we describe.
Methods
Sample collection
The study was approved under protocols at Mount Sinai Health System (MSHS) (IRB-18-00638/ STUDY-18-00627 and IRB-20-03276), Boston Children’s Hospital (04-09-113R), National Institute of Allergy and Infectious Disease (NIAID, NIH) (05-I-0213), Rockefeller University (JCA-0700 and XFK-0815), the French Ethics Committee Comité de Protection des Personnes, the French National Agency for Medicine and Health Product Safety and the Institut National de la Santé et de la Recherche Médicale (protocols C10-13 and C10-14). Patients were approached in person at the hospital or by email obtained through the NIH’s DS-Connect national registry (https://dsconnect.nih.gov). All patient data were deidentified. Written informed consent for all individuals in this study was provided in compliance with an institutional review board protocol. All samples from uninfected patients were drawn in the context of an outpatient routine visit and patients exhibited no signs of infection (fever, runny nose, cough, sore throat). Demographics and characteristics of individuals with DS are outlined in Extended Data Tables 1 and 2. For the six patients with SLE included in Fig. 3 and Extended Data Fig. 4, four had severe disease with renal involvement, one had moderate disease without renal involvement and one had mild disease with renal involvement that was resolved at time of sampling (Extended Data Table 3). Half of the patients had active disease and the other three donors were in remission on treatment. From each donor, blood was drawn into a cell preparation tube with sodium heparin (BD Vacutainer). Plasma was isolated from Ficoll separation and stored at −80 °C until use. Whole blood was either directly fixed using Proteomic Stabilizer PROT1 (SmartTube) and frozen at −80 °C, or fresh whole-blood samples were stained by adding 270 µl blood directly to a MDIPA tube containing the lyophilized antibody panel, mixing and incubating for 30 min at room temperature, followed by stabilization with PROT1 and freezing.
Determination of clinical immune dysfunction score
A clinical questionnaire was filled out by patients’ legal guardian or referring physician. One point was administered for each autoimmune manifestation or infectious episode. In cases in which an episode (such as pneumonia) was characterized as recurrent or reported more than three times, we capped the score for this category at three points. For example, if a patient had hypothyroidism and 12 ear infections, the total immune clinical score for this patient would be 4 points.
Mass cytometry
The detailed protocol for mass cytometry was previously described51. Frozen stabilized blood samples were thawed according to the manufacturer’s recommended protocol, then washed with barcode permeabilization buffer (Fluidigm). The samples were uniquely barcoded using the Cell-ID 20-Plex Pd Barcoding Kit (Fluidigm) and pooled together. For previously unstained samples, cells were incubated with an antibody cocktail for surface markers to identify major immune populations, followed by methanol permeabilization, heparin-block and stain with a cocktail of antibodies against intracellular targets, including markers of phosphorylation and signalling. All of the following antibodies were purchased from Fluidigm: Cd111-granzyme B, Cd112-IgA, In113-CD57, In115-CD11c, Cd116-IgD, I127-127I, Ce140-140Ce, Pr141-Ki67, Nd142-CD19, Nd143-CD45RA, Nd144-CD103, Nd145-CD4, Nd146-CD8, Sm147-pSTAT5, 150Nd-pSTAT5, Nd148-CD16, Sm149-CD127, Sm149-pSTAT6, Nd150-CD1c, Eu151-CD123, Sm152-CD66b, Eu153-pSTAT1, Sm154-ICOS, Gd155-CD27, Gd156-p38, 158Gd-pSTAT3, Tb159-pMAPKAP2, Gd160-CD14, Dy161-CD56, Dy162-TCRgd, Dy162-CD169, Dy163-CD172a/b, Dy164-CD69, Ho165-CD64, Ho165-STAT3, Er166-CD25, Er167-pERK1/2, Er168-CD3, Tm169-CD71, Tm169-STAT1, Er170-CD38, Yb171-CD95, Yb171-CD141, Yb172-CD39, Yb173-Tbet, Yb174-HLADR, Lu175-pS6, Yb176-CD54, Pr141-IFNγ, Nd144-CD141, 171Yb-CD141, Sm147-IL-1B, Sm149-IL-1RA, Eu153-TNF, Gd156-IL-6, Gd158-IL-2, Tb159-GM-CSF, Dy164-IL-17A, Ho165-CCL4, Er166-IL-10, Tm169-IFNα2b, Yb173-IL-8, Lu175-IL-29 and Yb176-CXCL10. After washing, cells were incubated in freshly diluted 2.4% formaldehyde containing 125 nM Ir Intercalator (Fluidigm), 0.02% saponin and 30 nM OsO4 (ACROS Organics) for 30 min at room temperature. Samples were then washed and acquired immediately.
For acquisition, samples were washed with PBS + 0.2% BSA, PBS and then CAS buffer (Fluidigm). The final solution in CAS buffer consisted of 1 million cells per ml and a 1/20 dilution of EQ beads (Fluidigm). After routine instrument optimization, samples were acquired at a rate of <300 events per second on a Helios mass cytometer (Fluidigm) with a modified wide-bore injector (Fluidigm).
FCS files of acquired events were normalized and concatenated with Fluidigm acquisition software, deconvoluted using a MATLAB-based debarcoding application and the resulting files were analysed using Cytobank. Cell events were identified as Ir191/193-positive and Ce140-negative events. Doublets were excluded on the basis of Mahalanobis distance and barcode separation and with the Gaussian parameters calculated using the Helios CyTOF software. Downstream data analysis was performed on Cytobank, by biaxial gating of immune populations according to a previously published gating scheme51 outlined in Supplementary Fig. 1. All cell frequency analyses were restricted to adult HCs and adults with DS (aged at least 18 years) unless otherwise indicated.
Flow cytometry
B cell phenotyping
Cryopreserved PBMCs were thawed and allowed to rest briefly in complete RPMI medium supplemented with 10% FBS, 1% penicillin–streptomycin and 1% GlutaMax. Cells were immunostained with antibodies in 0.5% BSA in PBS for 1 h, washed 3 times in 0.5% BSA in PBS for 1 h and acquired immediately. The following antibodies were used: CD19 APC-Cy7 (SJ25C1), CD27 FITC (M-T271), CD38 APC (HIT2), CD38 PE-Cy7 (HIT2), CD11c PE (B LY6), IgD BV421 (IA6), CD21 APC (Bu32) and anti-human 9G4 IgG APC (provided by J. Farmer).
pSTAT1 staining
The IgG fraction was isolated from HC and DS plasma using the Protein G HP SpinTrap (Sigma-Aldrich). THP-1 cells were incubated with the IgG fraction diluted 1:3 in complete RPMI with 10% FBS for 20 min or recombinant anti-IFNGR2 antibodies (2 μg ml−1, Thermo Fisher Scientific, PA5-47938) at 37 °C for 20 min followed by stimulation with IFNγ (10 ng ml−1) for 20 min at 37 °C. Cells were stained with Zombie Aqua Live-Dead (BioLegend) for 30 min on ice, then fixed/permeabilized in 90% ice-cold methanol and stained with anti-phospho-STAT1-PE (1:25, BD) for 1 h on ice. Flow cytometry was acquired on the BD LSR Fortessa II system, and data were analysed with FlowJo.
Whole-blood cytokine blocking experiments
Whole blood was incubated for 4 h with tofacitinib (500 nM, ApexBio) or tocilizumab (50 μg ml−1, Selleckchem) or vehicle controls (DMSO and human IgG control, 50 μg ml−1, BioLegend, respectively). For IFN-I blocking, whole blood was incubated with a combination of anti-IFNAR2 (2.5 μg ml−1 PBL Assay Science), anti-IFNα (0.2 μg ml−1, PBL 31110–1) and IFNβ (0.2 μg ml−1, PBL 31401-1) antibodies or vehicle control for 4 h. For IL-10 blocking, whole blood was incubated with a combination of anti-IL-10 (5 μg ml−1, BioLegend) and anti-IL-10R (5 μg ml−1, BioLegend) for 4 h. After blocking, whole blood was fixed using Proteomic Stabilizer PROT1 (1.4× blood volume, SmartTube) for 10 min at room temperature and frozen at −80 °C.
B cell stimulation
For plasmablast differentiation of CD11c+ cells (Fig. 4a), 2–5 × 104 naive (CD19+CD38−CD27−CD11c−) and CD11c+ (CD19+CD11c+) B cells were sort-purified from the PBMCs of healthy donors using the IMI5L sorter (BD) and rested overnight in complete RPMI. They were then stimulated with IL-21 (50 ng ml−1; BioLegend), BAFF (100 ng ml−1, BioLegend), IL-10 (250 ng ml−1, BioLegend), IL-2 (5 ng ml−1) and R848 (1 μg ml−1; Invivogen) with or without IFNγ diluted in complete RPMI. Cells were collected at day 4 and stained for flow cytometry.
For the cytokine-induced plasmablast differentiation of B cells (Fig. 4d,e and Extended Data Fig. 5a–f), 2–5 × 104 sorted naive or total B cells were sorted from healthy donor PBMCs using fluorescence-activated cell sorting, the Pan B Cell Isolation Kit II or the Naive B Cell Isolation Kit II (Miltenyi Biotech), as indicated. They were cultured for 3–6 days in complete RPMI supplemented with IL-21 (50 ng ml−1; BioLegend), BAFF (10 ng ml−1, BioLegend), IL-2 (50 ng ml−1, BioLegend), R848 (1 μg ml−1; Invivogen) with or without IL-10 (250 ng ml−1, BioLegend) and/or anti-human IgM (5 μg ml−1, Southern Biotech) as indicated, in addition to either (1) plasma from HC individuals or individuals with DS that was depleted from IgG using the Protein G HP SpinTrap (Sigma-Aldrich) at a final dilution of 1:4 or (2) exogenous addition of IL-6 (100 ng ml−1, BioLegend), IFNα2b (100 U ml−1, Merck), IL-4 (20 ng ml−1, BioLegend) and IFNγ (20 ng ml−1, BioLegend). At least two healthy donors and four individuals with DS were tested for each experiment. Cells were collected at indicated days and stained for flow cytometry.
For cytokine-blocking experiments, sorted naive B cells were cultured as described above in the presence of the following blocking antibodies: anti-IFNGR2 (2 μg ml−1, Thermo Fisher Scientific, PA5-47938), anti-IFNAR2 (2.5 μg ml−1 PBL Assay Science), anti-IFNα (0.2 μg ml−1, PBL 31110–1) and IFNβ (0.2 μg ml−1, PBL 31401-1), tocilizumab (50 μg ml−1, Selleckchem), adalimumab (2 μg ml−1, Selleckchem) or a combination. Cells were collected at indicated days and stained for flow cytometry.
T cell–B cell co-cultures
For co-cultures of in vitro polarized T cells (Fig. 4f–h and Extended Data Fig. 5g–i). CD4 T cells were purified using MACS (Miltenyi Biotec) from healthy donor PBMCs and cultured in the presence of plate-bound anti-CD3 (OKT3, 10 μg ml−1) and anti-CD28 (CD28.2, 5 μg ml−1) antibodies in complete RPMI alone (unstimulated control) or with (1) IL-2 (50 ng ml−1), IL-12 (5 ng ml−1) and anti-IL-4 (1 μg ml−1, R&D MAB204-SP) (TH1 conditions); (2) IL-6 (50 ng ml−1) with or without IL-2 (50 ng ml−1); or (3) plasma from HC individuals or individuals with DS (1:2 ratio). After 4 days, T cells were washed and irradiated (5,000 rad) and co-cultured with freshly MACS-sorted naive B cells from the same donor. Cells were co-cultured for 3–6 days in complete RPMI supplemented with IL-21 (50 ng ml−1), IL-2 (50 ng ml−1) and anti-human IgM (5 μg ml−1, Southern Biotech) and collected for flow cytometry. The supernatants were collected for IFNγ ELISA quantification (BioLegend) according to the manufacturer’s instructions.
For T cell–B cell co-cultures from HC donors and donors with DS (Fig. 4i,j), CD4 T cells were MACS-purified and co-incubated with sort-purified CD19+CD38−CD27−CD11c− B cells on the same day. B and T cells were co-cultured for 3 days in complete RPMI supplemented with IL-21, IL-2 and anti-human IgM as above and collected for flow cytometry.
BCR analysis
Sample preparation
Donor PBMCs were thawed in complete RPMI and rested overnight. Naive (CD19+CD27−CD38lowCD11c−), CD11c+ (CD19+CD27−CD38lowCD11c−) and memory (CD19+CD27+CD38−) B cells were sort-purified using an IMI5L sorter (BD) and genomic DNA was isolated with QIAamp DNA Blood Mini Kit (Qiagen).
Library preparation
Sample data were generated using the immunoSEQ Assay (Adaptive Biotechnologies). The somatically rearranged Homo sapiens BCR IgH locus CDR3 was amplified from genomic DNA using a two-step, amplification-bias-controlled multiplex PCR approach52,53. The first PCR consists of forward and reverse amplification primers specific for every V and J gene segment and amplifies the hypervariable complementarity-determining region 3 (CDR3) of the immune receptor locus. Furthermore, primers are included to amplify B cell precursor DJ rearrangements. The second PCR adds a proprietary barcode sequence and Illumina adapter sequences54. CDR3 libraries were sequenced on the Illumina instrument according to the manufacturer’s instructions.
Data analysis
Raw sequencing reads were demultiplexed according to Adaptive’s proprietary barcode sequences. Demultiplexed reads were then processed to remove adapter and primer sequences, identify and remove primer dimer, germline and other contaminant sequences. The filtered data are clustered using the relative frequency ratio between similar clones and a modified nearest-neighbour algorithm to merge closely related sequences to correct technical errors introduced through PCR and sequencing while allowing for somatic hypermutation in the V segment. The resulting sequences were sufficient to allow annotation of the V, D and J genes, and the N1 and N2 regions constituting each unique CDR3 and the translation of the encoded CDR3 amino acid sequence. Gene definitions were based on annotation in accordance with the IMGT database (www.imgt.org). The set of observed biological BCR IgH CDR3 sequences was normalized to correct for residual multiplex PCR amplification bias and quantified against a set of synthetic BCR IgH CDR3 sequence analogues53. Data were analysed using the immunoSEQ Analyzer toolset.
Multiplex cytokine analysis
Plasma collected by Ficoll isolation from heparinized whole blood was clarified by centrifugation. Magnetic Luminex assays with the MILLIPLEX MAP Human Cytokine/Chemokine Magnetic Bead Panel (MilliporeSigma, HCYTMAG-60 K-PX30) were run according to the manufacturer’s protocol. The samples were quantified on the MAGPIX xMAP Instrument (Luminex). For each sample, >50 beads were collected per analyte. The median fluorescence intensity of these beads was recorded and used for analysis with the Milliplex Analyst software using a 5P regression algorithm. In the absence of signal, the assay’s lower limit of detection for each cytokine was used. Patients with DS aged 7 months to 37 years (n = 21) and control individuals aged 6 years to 39 years (n = 10) were included.
Auto-antibody analysis
Seromic profiling of autoantibodies was conducted as previously described55. The CDI HuProt peptide array was run according to the manufacturer’s instructions, at 1/500 to avoid low-tittered cross-reactivity and using a robust blocking buffer to prevent unspecific binding. After applying patient sera, chips were washed and secondary fluorescent antibodies to quantify IgG (Cy3; 532 nm) and IgA (Cy5; 635 nm) antibody isotypes were applied at the manufacturer’s recommended concentrations. Each spot on the array, representing a protein printed in duplicate, was gated using Genepix software alignment and then manually quality controlled to ensure proper quantification and removal of possible artifacts. High differences between replicate spots (CV > 0.5) were flagged, along with information about staining artifacts (rare and excluded). Raw sample-peptide matrices were then read into the R (v.4.0.4) statistical environment to be processed by the limma microarray analysis suite (v.3.46.0)56. On a per-sample and a per-isotype (IgG or IgA) basis, the background intensity was subtracted using the backgroundCorrect function (method: normexp, normexp.method: rma) and then normalized across all samples using the normalizeBetweenArrays function (method=“cyclicloess”). Next, all control and non-detected peptides were filtered from the sample–peptide matrices and duplicate spot intensities were averaged across all of the remaining peptides on a per-sample basis. Individually processed sample matrices were concatenated, column-wise, to generate an experimental matrix containing all samples and peptides shared across all arrays and to be analysed for differential peptide reactivity using the limma differential expression functions. First, to exclude low-reactive peptides and outlier reactivity, peptides were filtered to include only peptides that had reactivities above the median signal intensity in at least 5 samples (of the 50 total samples processed). PCA was then performed using the base R (v.4.0.4) prcomp function (scale: TRUE) and PCA and loadings were visualized using the fviz_pca_ind function from the FactoMiner/factoextra package (v.1.0.7)57. To determine differentially abundant autoantibodies, we first fit a linear model for each peptide to model the variation of peptide reactivity (against an auto-antibody) across all arrays using limma’s lmFit function. After building the model, we generated a contrast matrix to test disease groups against healthy controls. Differential autoantibody abundance testing was performed and moderated under the empirical Bayes framework described by the developer and resulting peptide lists were filtered against an absolute log2[fold change] > 1 and a P < 0.05. Overlap analyses were conducted and visualized using the UpSetR (v.1.4.0)58 and ggvennDiagram (https://github.com/gaospecial/ggVennDiagram) (v.1.2.1) packages. Heat maps were generated using the ComplexHeatmap (v.2.7.4)59 library.
Chromosomal reactivity heatmap
To assess whether autoantibody reactivity was influenced by the gene dosage effect in DS, filtered differentially abundant autoantibodies (obtained from the DS versus HC contrast; see Supplementary Data 1) were plotted across the 23 human chromosomes (hg38) in a location-dependent manner. In brief, gene names were extracted from their associated peptides and chromosomal locations were identified using the org.Hs.eg.db (v.3.12.0)60 database. Hits were further filtered against a fold-change of 1.5 for plotting purposes and the karyoplotR (v.1.16.0)61 package was then used for visualization.
Tissue-protein expression heat map
Tissue-protein detection values of all human proteins were extracted from the Human Protein Atlas API using the HPAanalyze (v.1.8.1)62 package. Differentially abundant autoantibodies (obtained from the DS versus HC contrast; Supplementary Data 1) were subset from the full dataset (Supplementary Data 2) and visualized using the ComplexHeatmap library.
GO analysis
GO enrichment analysis was conducted on over-abundant (log2[fold change] > 1) autoantibodies with limma’s goana function, and biological process GO terms with P < 0.0001 were subset and visualized using the ggplot263 (v.3.3.3) package.
ELISA assays
Recombinant protein ELISA
Overnight, 96-well Costar plates were coated at 4 °C with 100 μl per well of a 1 μg ml−1 solution of recombinant IFNGR2, MSTN or ATP6V1G2 protein (OriGene) suspended in 1× PBS. The next morning, the coating solution was removed and wells were washed three times with 100 μl of washing buffer (PBS with 0.05% (v/v) Tween 20; PBS-T). Next, 200 μl of blocking buffer (PBS with 1% with bovine plasma albumin (endotoxin-free)) was added to each well at room temperature and incubated at room temperature for 1 h. Plasma samples were diluted 1:100 in blocking buffer.
Total IgG and 9G4 IgG ELISA
Overnight, 96-well Costar plates were coated at 4 °C with 100 μl per well of a 2 μg ml−1 solution of goat anti-human IgG, F(ab′)2-fragment-specific (Jackson ImmunoResearch) or 1:100 dilution of anti-human 9G4 IgG antibody (provided by J. Farmer) suspended in 1× PBS. The next morning, the coating solution was removed and wells were washed with three times with 100 μl of washing buffer (PBS with 0.05% (v/v) Tween-20; PBS-T). Next, 200 μl of blocking buffer (PBS with 1% with bovine plasma albumin (endotoxin-free)) was added to each well at room temperature and incubated at room temperature for 1 h. The supernatants from B cell stimulation experiments were diluted 1:10.
Antibody detection
Plates were then washed three times before adding goat anti-human IgG F(ab)–horseradish peroxidase (HRP)-conjugated secondary antibody (diluted 1:3,000 in PBS-T). The plates were incubated for 1 h at 37 °C and washed three times. The plates were again washed three times with wash buffer. Once completely dry, 100 μl SIGMAFAST OPD (o-phenylenediamine dihydrochloride; Sigma–Aldrich) solution was added to each well. This substrate was left on the plates for 5 min and the reaction was then stopped by the addition of 50 μl per well of 3 M hydrochloric acid. The optical density at 490 nm was measured using the POLARstar Omega (BMG Labtech) plate reader.
Detection of anti-type I IFN autoantibodies
Detection of anti-type I IFN autoantibodies was performed using Gyros as described previously64. Cytokines, recombinant human IFNα2 (Miltenyi Biotec, 130-108-984) or recombinant human IFNω (Merck, SRP3061) were first biotinylated with EZ-Link Sulfo-NHS-LC-Biotin (Thermo Fisher Scientific, A39257), according to the manufacturer’s instructions, with a biotin-to-protein molar ratio of 1:12. The detection reagent contained a secondary antibody (Alexa Fluor 647 goat anti-human IgG (Thermo Fisher Scientific, A21445)) diluted in Rexxip F (Gyros Protein Technologies, P0004825; 1:500 dilution of the 2 mg ml−1 stock to yield a final concentration of 4 μg ml−1). Phosphate-buffered saline, 0.01% Tween-20 (PBS-T) and Gyros Wash buffer (Gyros Protein Technologies, P0020087) were prepared according to the manufacturer’s instructions. Plasma or serum samples were then diluted 1:100 in 0.01% PBS-T and tested with the Bioaffy 1000 CD (Gyros Protein Technologies, P0004253) and the Gyrolab xPand (Gyros Protein Technologies, P0020520). Cleaning cycles were performed in 20% ethanol.
Functional evaluation of anti-type I IFNs autoantibodies using luciferase reporter assays
The blocking activity of anti-IFNα2 and anti-IFNω autoantibodies was determined with a reporter luciferase activity as described previously64. HEK293T cells were transfected with a plasmid containing the firefly luciferase gene under the control of the human ISRE promoter in the pGL4.45 backbone and a plasmid constitutively expressing Renilla luciferase for normalization (pRL-SV40). Cells were transfected in the presence of the X-tremeGENE9 transfection reagent (Sigma-Aldrich, 6365779001) for 24 h. Cells in Dulbecco’s modified Eagle medium (DMEM; Thermo Fisher Scientific) supplemented with 2% fetal calf serum and 10% healthy control or patient serum/plasma (after inactivation at 56 °C for 20 min) were stimulated with IFNα2 (Miltenyi Biotec, 130-108-984) and IFNω (Merck, SRP3061) at 10 ng ml−1 or 100 pg ml−1 for 16 h at 37 °C. Each sample was tested once for each cytokine and dose. Finally, cells were lysed for 20 min at room temperature, and luciferase levels were measured with the Dual-Luciferase Reporter 1000 Assay System (Promega, E1980) according to the manufacturer’s protocol. Luminescence intensity was measured with a VICTOR-X Multilabel Plate Reader (PerkinElmer Life Sciences). Firefly luciferase activity values were normalized against Renilla luciferase activity values.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The data supporting the findings of this study are available in the Article and its Supplementary Information.
References
Bull, M. J. Down syndrome. N. Engl. J. Med. 382, 2344–2352 (2020).
Ryan, C., Vellody, K., Belazarian, L. & Rork, J. F. Dermatologic conditions in Down syndrome. Pediatr. Dermatol. 38, 14731 (2021).
Down, J. L. H. Observations on an ethnic classification of idiots. Lond. Hosp. Rep. 3, 259–262 (1866).
Mai, C. T. et al. National population-based estimates for major birth defects, 2010–2014. Birth Defects Res. 111, 1420–1435 (2019).
Antonarakis, S. E. et al. Down syndrome. Nat. Rev. Dis. Prim. 6, 9 (2020).
Rivelli, A. et al. Prevalence of endocrine disorders among 6078 individuals with Down syndrome in the United States. J. Patient Cent. Res. Rev. 9, 70–74 (2022).
Kong, X.-F. et al. Three copies of four interferon receptor genes underlie a mild type I interferonopathy in Down syndrome. J. Clin. Immunol. 40, 807–819 (2020).
Sullivan, K. D. et al. Trisomy 21 consistently activates the interferon response. eLife 5, e16220 (2016).
Malle, L. et al. Excessive negative regulation of type I interferon disrupts viral control in individuals with Down syndrome. Immunity 55, 2074–2084 (2022).
Giménez-Barcons, M. et al. Autoimmune predisposition in Down syndrome may result from a partial central tolerance failure due to insufficient intrathymic expression of AIRE and peripheral antigens. J. Immunol. 193, 3872–3879 (2014).
Araya, P. et al. Trisomy 21 dysregulates T cell lineages toward an autoimmunity-prone state associated with interferon hyperactivity. Proc. Natl Acad. Sci. USA 116, 24231–24241 (2019).
Cossarizza, A. et al. Age-related expansion of functionally inefficient cells with markers of natural killer activity in Down’s syndrome. Blood 77, 1263–1270 (1991).
Verstegen, R. H. J. & Kusters, M. A. A. Inborn errors of adaptive immunity in Down syndrome. J. Clin. Immunol. 40, 791–806 (2020).
Taft, J. & Bogunovic, D. The Goldilocks zone of type I IFNs: lessons from human genetics. J. Immunol. 201, 3479–3485 (2018).
Rea, I. M. et al. Age and age-related diseases: role of inflammation triggers and cytokines. Front. Immunol. 9, 586 (2018).
Zhang, Y. et al. Aberrations in circulating inflammatory cytokine levels in patients with Down syndrome: a meta-analysis. Oncotarget 8, 84489–84496 (2017).
Huggard, D. et al. Increased systemic inflammation in children with Down syndrome. Cytokine 127, 154938 (2020).
Junttila, I. S. Tuning the cytokine responses: an update on interleukin (IL)-4 and IL-13 receptor complexes. Front. Immunol. 9, 888 (2018).
Lambert, K. et al. Deep immune phenotyping reveals similarities between aging, Down syndrome, and autoimmunity. Sci. Transl. Med. 14, 4888 (2022).
Milner, J. D. et al. Early-onset lymphoproliferation and autoimmunity caused by germline STAT3 gain-of-function mutations key points. Blood 21, 25 (2015).
Fabre, A. et al. Clinical aspects of STAT3 gain-of-function germline mutations: a systematic review. J. Allergy Clin. Immunol. Pract. 7, 1958–1969 (2019).
Verstegen, R. H. J. et al. Defective B-cell memory in patients with Down syndrome. J. Allergy Clin. Immunol. 134, 1346–1353 (2014).
Dieudonné, Y. et al. Immune Defect in adults with down syndrome: insights into a complex issue. Front. Immunol. 11, 840 (2020).
Song, W. et al. Development of Tbet- and CD11c-expressing B cells in a viral infection requires T follicular helper cells outside of germinal centers. Immunity 55, 290–307 (2022).
Karnell, J. L. et al. Role of CD11c+ T-bet+ B cells in human health and disease. Cell. Immunol. 321, 40–45 (2017).
Woodruff, M. C. et al. Extrafollicular B cell responses correlate with neutralizing antibodies and morbidity in COVID-19. Nat. Immunol. 21, 1506–1516 (2020).
Jenks, S. A. et al. Distinct effector B cells induced by unregulated toll-like receptor 7 contribute to pathogenic responses in systemic lupus erythematosus. Immunity 49, 725–739 (2018).
Keller, B. et al. The expansion of human T-bethighCD21low B cells is T cell dependent. Sci. Immunol. 6, 52 (2021).
Li, H., Borrego, F., Nagata, S. & Tolnay, M. Fc receptor-like 5 expression distinguishes two distinct subsets of human circulating tissue-like memory B cells. J. Immunol. 196, 4064–4074 (2016).
Sokol, C. L. & Luster, A. D. The chemokine system in innate immunity. Cold Spring Harb. Perspect. Biol. 7, a016303 (2015).
Vestergaard, C. et al. A Th2 chemokine, TARC, produced by keratinocytes may recruit CLA+CCR4+ lymphocytes into lesional atopic dermatitis skin. J. Invest. Dermatol. 115, 640–646 (2000).
Mikhak, Z., Strassner, J. P. & Luster, A. D. Lung dendritic cells imprint T cell lung homing and promote lung immunity through the chemokine receptor CCR4. J. Exp. Med. 210, 1855 (2013).
Zumaquero, E. et al. IFNγ induces epigenetic programming of human T-bethi B cells and promotes TLR7/8 and IL-21 induced differentiation. eLife 8, e41641 (2019).
Wang, S. et al. IL-21 drives expansion and plasma cell differentiation of autoreactive CD11chi T-bet + B cells in SLE. Nat. Commun. 9, 1758 (2018).
Jego, G. et al. Plasmacytoid dendritic cells induce plasma cell differentiation through type I interferon and interleukin 6. Immunity 19, 225–234 (2003).
Naradikian, M. S. et al. Cutting edge: IL-4, IL-21, and IFN-γ interact to govern T-bet and CD11c expression in TLR-activated B cells. J. Immunol. 197, 1023–1028 (2016).
Stone, S. L. et al. T-bet transcription factor promotes antibody-secreting cell differentiation by limiting the inflammatory effects of IFN-γ on B cells. Immunity 50, 1172–1187 (2019).
Bashford-Rogers, R. J. M. et al. Analysis of the B cell receptor repertoire in six immune-mediated diseases. Nature 574, 122–126 (2019).
Tipton, C. M., Hom, J. R., Fucile, C. F., Rosenberg, A. F. & Sanz, I. Understanding B-cell activation and autoantibody repertoire selection in systemic lupus erythematosus: a B-cell immunomics approach. Immunol. Rev. 284, 120–131 (2018).
Richardson, C. et al. Molecular basis of 9G4 B cell autoreactivity in human systemic lupus erythematosus. J. Immunol. 191, 4926–4939 (2013).
Marcovecchio, G. E. et al. Thymic epithelium abnormalities in DiGeorge and Down syndrome patients contribute to dysregulation in T cell development. Front. Immunol. 10, 447 (2019).
Pierce, M. J., LaFranchi, S. H. & Pinter, J. D. Characterization of thyroid abnormalities in a large cohort of children with Down syndrome. Horm. Res. Paediatr. 87, 170 (2017).
Mader, S., Brimberg, L. & Diamond, B. The role of brain-reactive autoantibodies in brain pathology and cognitive impairment. Front. Immunol. 8, 1101 (2017).
Lepagnol-Bestel, A. M. et al. DYRK1A interacts with the REST/NRSF-SWI/SNF chromatin remodelling complex to deregulate gene clusters involved in the neuronal phenotypic traits of Down syndrome. Hum. Mol. Genet. 18, 1405–1414 (2009).
van der Poel, C. E., Spaapen, R. M., van de Winkel, J. G. J. & Leusen, J. H. W. Functional characteristics of the high affinity IgG receptor, FcγRI. J. Immunol. 186, 2699–2704 (2011).
Bustamante, J., Boisson-Dupuis, S., Abel, L. & Casanova, J. L. Mendelian susceptibility to mycobacterial disease: genetic, immunological, and clinical features of inborn errors of IFN-γ immunity. Semin. Immunol. 26, 454–470 (2014).
Malle, L. et al. Individuals with Down syndrome hospitalized with COVID-19 have more severe disease. Genet. Med. 23, 576–580 (2020).
Gillespie, K. M. et al. Islet autoimmunity in children with Down’s syndrome. Diabetes 55, 3185–3188 (2006).
Kohen, D. & Wise, P. H. Autoantibodies in Down’s syndrome. Lancet 340, 430 (1992).
Cardinale, K. M. et al. Immunotherapy in selected patients with Down syndrome disintegrative disorder. Dev. Med. Child Neurol. 61, 847–851 (2019).
Geanon D, et al. A streamlined whole blood CyTOF workflow defines a circulating immune cell signature of COVID-19. Cytometry A. 99, 446-461 (2021).
Robins, H. S. et al. Comprehensive assessment of T-cell receptor β-chain diversity in αβ T cells. Blood 114, 4099–4107 (2009).
Carlson, C. S. et al. Using synthetic templates to design an unbiased multiplex PCR assay. Nat. Commun. 4, 2680 (2013).
Robins, H. et al. Ultra-sensitive detection of rare T cell clones. J. Immunol. Methods 375, 14–19 (2012).
Gnjatic, S. et al. Seromic profiling of ovarian and pancreatic cancer. Proc. Natl Acad. Sci. USA 107, 5088–5093 (2010).
Ritchie, M. E. et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47 (2015).
Lê, S., Josse, J. & Husson, F. FactoMineR: an R package for multivariate analysis. J. Stat. Softw. 25, 1–18 (2008).
Conway, J. R., Lex, A. & Gehlenborg, N. UpSetR: an R package for the visualization of intersecting sets and their properties. Bioinformatics 33, 2938–2940 (2017).
Gu, Z., Eils, R. & Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 32, 2847–2849 (2016).
Carlson, M. org.Hs.eg.db (2017).
Gel, B. karyoploteR (2017).
Nhat, A. HPAanalyze (2018).
Wickham, H. ggplot2 (2009); https://doi.org/10.1007/978-0-387-98141-3.
Bastard, P. et al. Autoantibodies neutralizing type I IFNs are present in ~4% of uninfected individuals over 70 years old and account for ~20% of COVID-19 deaths. Sci. Immunol. 6, abl4340 (2021).
Acknowledgements
We thank all of the patients and their families for their participation; A. Rahman, D. Geanon and G. Kelly from the Human Immune Monitoring Centre at the Icahn School of Medicine for their technical assistance; and J. Farmer and K. Hillier for sharing reagents. This study was funded by the National Institute of Allergy and Infectious Diseases Grants R01AI150300, R01AI150300-01S1 and R01AI151029. L. Malle was supported by the National Institute of Child Health and Human Development T32 training grant T32HD075735. L.N. is supported by the Division of Intramural Research, National Institute of Allergy and Infectious Diseases, NIH. S.G. was supported by NIH grants CA224319, DK124165 and CA196521.
Author information
Authors and Affiliations
Contributions
L. Malle designed and performed experiments and analysed the data for cytokine array, CyTOF, B–T cell co-cultures and BCR sequencing, and wrote the manuscript. R.S.P. analysed the CDI array and edited the manuscript. M.M.-F. performed steady-state B–T cell co-cultures from DS donors. O.S. performed 9G4 and IFNy ELISA. J.T. edited the manuscript. Q.P. performed IFN autoantibody Gyros and neutralization assays. S.B. and A.R. coordinated DS cohort and processed whole-blood samples. V.B., K.T. and S.G. performed the CDI array. B.R.R. helped to analyse BCR sequencing data. P.B., J.S., C.M., A.-S.R., L. Maillebouis, M.V.-M., R.T., J.-L.C., L.N. and D. Bush recruited patients. D. Bogunovic supervised the work, wrote the manuscript, and helped to design the experiments and analyse the data.
Corresponding author
Ethics declarations
Competing interests
D.B. is the founder and part owner of Lab11 Therapeutics. S.G. reports other research funding from Genentech, Boehringer-Ingelheim, Celgene, Takeda, and Regeneron.
Peer review
Peer review information
Nature thanks Stuart Tangye and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 Global cytokine dysregulation in DS.
(A-B) Correlation between cytokine group and (A) Age and (B) Clinical Immune Dysfunction score (determined according to reported clinical history, see Methods) and Age in individuals with DS. Significance assessed by one-way ANOVA with Tukey’s post-hoc analysis, ns denotes p > 0.05, *p ≤ 0.05; **p ≤ 0.005, ***p ≤ 0.0005. (C) Multiplex cytokine analysis by Magnetic Luminex assay of plasma from HCs (n = 10), uninfected individuals with DS (n = 21), and individual with acute respiratory infection (n = 1), or individuals with DS and COVID-19 (n = 7) with severity and time of sampling as indicated, expressed as log2FC over the mean HC per cytokine. Unsupervised clustering of samples and cytokines using the complete method (distance metric: Euclidean). (D) Intracellular staining of IL-6 in CD4 T cells, CD8 T cells, and Myeloid cells from HCs (n = 4) and individuals with DS (n = 5).
Extended Data Fig. 2 Cellular immune landscape in DS.
(A) Representative t-SNE of agranulocytes adults with DS (n = 3) and age-matched HCs (n = 3) illustrating the immune cell distribution in whole blood. (B-E) Frequencies of (B) agranulocyte subsets, (C) granulocyte subsets, and (D) CD4 and (E) CD8 T cell subsets in whole blood from adults with DS (n = 10), adults with DS and COVID-19 (n = 5), and age-matched HCs (n = 8). (F-G) Frequencies (F) and absolute counts (G) of T regulatory (Treg) cells (CD4+CD25+CD127−) and naive (CD45RA+) and memory (CD45RA−) subsets in whole blood from adults with DS (n = 5) and age-matched HCs (n = 5). (H-I) Representative plots and calculated frequencies of (H) T helper (Th) and (I) T follicular helper (Tfh) cell subsets in whole blood from adults with DS (n = 5) and age-matched HCs (n = 5), expressed as percent of memory CD4 T cells. In (B-E), whiskers denote min and max values, bounds of box denote Q1–Q3, and centre bar denotes mean. (F-I) Error bars denote SD. (B-E) Significance assessed by one-way ANOVA with Tukey’s post-hoc analysis, ns denotes p > 0.05, *p ≤ 0.05; **p ≤ 0.005, ***p ≤ 0.0005. (F-I) Significance assessed by two-tailed paired t tests, ns denotes p > 0.05, *p ≤ 0.05; **p ≤ 0.005, ***p ≤ 0.0005.
Extended Data Fig. 3 Basal signalling in CD4 T cells in DS.
(A) Basal STAT3 phosphorylation in CD4 T cell subsets from HCs (n = 13), individuals with DS (n = 19), and adults with DS and COVID-19 (n = 5) expressed as Log2FC over the mean HCs per subset. Significance assessed by one-way ANOVA with Tukey’s post-hoc analysis, ns denotes p > 0.05, *p ≤ 0.05; **p ≤ 0.005, ***p ≤ 0.0005. Whiskers denote min and max values, bounds of box denote Q1–Q3, and centre bar denotes mean. (B) Basal STAT5 phosphorylation in CD4 T cell subsets from individuals with DS (n = 14) and age-matched controls (n = 10), expressed as Log2FC over the mean HCs per subset. (C-D) STAT3 phosphorylation in CD4 T cell subsets expressed as Log2FC over the mean mock-treated HCs per subset after ex vivo whole blood treatment for 4 h with (C) Tofacitinib (500nM) (n = 6 HC, n = 7 DS), (D) Tocilizumab (50 μg ml−1) (n = 4 HC, n = 7 DS). (E) Surface expression of IL-6R in CD4 T cells from adults with DS (n = 3) and age-matched HCs (n = 3). (F) STAT3 phosphorylation in CD4 T cell subsets induced by stimulation of whole blood with recombinant IL-6 (50 ng ml−1) for 15 min, expressed as Log2FC over the mean mock-treated HCs (n = 2 HC, n = 4 DS). Box plots denote min and max values for HCs, error bars indicate SD and centre denotes mean for DS. (C-E) Error bars denote SD. (B-F) Significance assessed by two-tailed paired t tests, ns denotes p > 0.05, *p ≤ 0.05; **p ≤ 0.005, ***p ≤ 0.0005.
Extended Data Fig. 4 Abnormal B cell subsets in DS.
(A) Raw B cell counts in HCs (n = 6) and adults with DS (n = 7) or patients with SLE (n = 4). Error bars denote SD. (B) Gating scheme of B cells subtypes. (C) Frequency in children with DS (n = 11) and age-matched HCs (n = 7) of total B cells expressed as percent of CD66b− cells (non-granulocytes), Significance assessed by two-tailed unpaired t tests, *p ≤ 0.05. (D-F) Frequency in children with DS (n = 8) and age-matched HCs (n = 4) of CD11c+ B cells in the IgD+ naive or DN compartments, expressed as percent of (D) total, (E) IgD+ naïve, or (F) DN B cells. (G) Ratios of CD11c+ subsets to CD11c− subset (rN:aN and DN2:DN1) in HC and DS groups, Log2-transformed. Significance assessed by two-tailed unpaired t tests, *p ≤ 0.05. (H-I) Correlation of CD11c+ B cell frequency and (H) age and (I) total cytokines (calculated as the sum of all circulating cytokines, pg ml−1) in individuals (H) or adults (I) with DS and age-matched controls. (r: Pearson correlation coefficient). (J) Correlation of CD11c+ B cell frequency and autoimmune score (calculated as the sum of autoimmune diseases) in individuals with DS. (r: Pearson correlation coefficient). (K) Total IgG in the plasma of HCs (n = 7) and individuals with DS (n = 12) or SLE (n = 6) assessed by ELISA. Significance assessed by one-way ANOVA with Tukey’s post-hoc analysis, ns denotes p > 0.05, *p ≤ 0.05. In (C-F, K), whiskers denote min and max values, bounds of box denote Q1–Q3, and centre bar denotes mean.
Extended Data Fig. 5 Atypical activation of naive B cells by cytokines and activated T cells.
(A Plasmablast differentiation of MACS-isolated total B cells from 2 healthy donors after 3-day culture in the presence of BAFF (10 ng ml−1), IL-2 (50 ng ml−1), IL-21 (50 ng ml−1), R848 (1 μg ml−1), anti-IgM (1 μg ml−1), and IgG-depleted plasma from HCs (n = 3) or individuals with DS (n = 3), run in duplicates. (B-C) MACS-isolated total B cells from 2 healthy donors were cultured in the presence of BAFF, IL-2, IL-21, R848, anti-IgM (same concentrations as A), in the presence of IL-6 (100 ng ml−1), IFN-α2b (100 U ml−1), or both, run in duplicates. After 3 days, cells were washed and cultured for another 3 days in media with anti-IgM. (B) Plasmablast differentiation at day 3 and (C) ELISA for total IgG in supernatant at day 6. (D-E) MACS-isolated total B cells from 2 healthy donors were cultured for 3-days in the presence of BAFF, IL-2, IL-21, R848, anti-IgM (same concentrations as A), and IL-4 (20 ng ml−1), IFN-ɣ (20 ng ml−1), or both, run in duplicates. Cells were then washed and cultured for another 3 days in media with anti-IgM. (D) Plasmablast differentiation at day 3 and (E) ELISA for total IgG in supernatant at day 6. (F) Plasmablast differentiation of MACS-isolated naive B cells from a healthy donor after 3-day culture in the presence of BAFF, IL-2, IL-21, R848, anti-IgM (same concentrations as J), and IgG-depleted DS plasma in the presence of Tofacitinib (500nM) or antibodies blocking IFN-I, IFN-II, IL-6, and TFN-ɑ signalling, run in duplicates. (G-H) Co-cultures containing T cells activated with IL-6, IL-2, both, or polarized into “Th1 cells” together with MACS-isolated naive B cells from the same donor, run in triplicates. (G) Intracellular T-bet expression in non-plasmablast B cells and (H) quantification of IFN-g in the supernatant after 3-6 days of co-culture. (I) Frequency of plasmablasts in co-cultures containing T cells previously polarized with serum from HCs (n = 3) or individuals with DS (n = 3) together with MACS-isolated naive B cells from the same donor. Significance assessed by two-tailed unpaired t-test. (A-I) Error bars denote SD. (A,H) Significance assessed by One-way ANOVA with Tukey’s post-hoc analysis, ns denotes p > 0.05; **p ≤ 0.005; ***p ≤ 0.0005; ****p ≤ 0.0001.
Extended Data Fig. 6 Receptor sequencing in atypical B cells.
(A) Expression of IgD and IgA in CD11c+ B cells from adults with DS (n = 3) and age-matched HCs (n = 3). Significance assessed by unpaired t-tests, ns denotes p > 0.05. Error bars denote SD. (B-F) BCR sequencing from gDNA isolated from sorted naïve, CD11c+ and memory B cells from controls (n = 6) and individuals with DS (n = 6). (B) Sorting scheme for naïve, CD11c+ and memory B cells (left) and number of cells sorted and number of productive BCRs obtained by sequencing for each cell type (right). (C) Fraction of in-frame BCRs containing no stop codons (“productive BCRs”) in naïve, CD11c+ and memory B cells from controls (n = 6) and individuals with DS (n = 6). (D) Simpson clonality in productive BCRs from CD11c+ B cells from controls (n = 6) and individuals with DS (n = 6). In (C-D), significance assessed by two-tailed unpaired t-test (ns denotes p > 0.05) and whiskers denote min and max values, bounds of box denote Q1–Q3, and centre bar denotes mean. (E-F) Representative heatmaps of Morisita index indicating overlap between B cell subsets in HC and DS at the (E) nucleotide and (F) amino acid levels. (G) Heatmap of IGHV gene frequency in HC and DS. Only genes that occurred at higher than 0.1% frequency in more than 10 samples are shown. (H) 9G4 IgG antibodies in supernatant after 4-day culture of sorted HC naïve, CD11c+ or memory B cells in the presence of BAFF, IL-2, IL-10, IL-21, the TLR7/8 ligand R848 with or without IFN-ɣ. Results representative of 2 independent experiments.
Extended Data Fig. 7 Autoantibodies in DS plasma.
(A) Principal component analysis of HuProt IgA dataset for adult HCs (n = 4), adults with DS (n = 5), and patients, Immunodysregulation polyendocrinopathy enteropathy X-linked syndrome (IPEX) (n = 3) and Autoimmune polyglandular syndrome type 1 (APS-1) (n = 1). (B) Number of IgA autoantigens enriched at least 2-fold in DS, IPEX, and APS-1 compared to HCs. (C) Venn diagram of enriched IgA autoantigens overlapping between disease groups. (D) Heatmap of enriched IgA autoantigens in HCs, DS, IPEX, and APS-1. Colour intensity corresponds to the log2FC expression value relative to the mean of healthy adult controls. (E) Component loadings (PC1 and PC2) from Principal component analysis of HuProt IgG dataset for adults with DS (n = 5) and age-matched HCs (n = 4). (F) ELISA of anti-MSTN and anti-ATP6V1G2 autoantibodies in plasma from HCs (n = 6) and individuals with DS (n = 6). Significance assessed by two-tailed unpaired t tests, * denotes p ≤ 0.05. Whiskers denote min and max values, bounds of box denote Q1–Q3, and centre bar denotes mean. (G) Heatmap of IgG type I IFN autoantigens in HCs, DS, IPEX, and APS-1. Colour intensity corresponds to the log2FC expression value relative to the mean of healthy adult controls. (H) Quantification of antibodies against IFNα2 and IFNω in serum from HCs (n = 32), DS without COVID-19 (n = 10), DS with COVID-19 (n = 6), and APS-1 (n = 1) by Gyros assay. (I) Neutralization of IFNα2 and IFNω activity by serum from HCs (n = 32), DS without COVID-19 (n = 10), DS with COVID-19 (n = 6), and APS-1 (n = 1) by Gyros assay. (H-I) Significance assessed by One-way ANOVA with Tukey’s post-hoc analysis, ns denotes p > 0.05, *p ≤ 0.05; **p ≤ 0.005; ***p ≤ 0.0005; ****p ≤ 0.0001.
Extended Data Fig. 8 Graphical abstract.
Breaking Immune Tolerance in Down Syndrome: A Triad of Cytokines, Activated T cells and CD11c+ B Cells. Created with BioRender.
Supplementary information
Supplementary Figure 1
Schematic of the gating hierarchy used to define immune populations by mass cytometry.
Supplementary Data 1
Differential peptide analysis of HuProt IgG dataset.
Supplementary Data 2
HuProt raw data files.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Malle, L., Patel, R.S., Martin-Fernandez, M. et al. Autoimmunity in Down’s syndrome via cytokines, CD4 T cells and CD11c+ B cells. Nature 615, 305–314 (2023). https://doi.org/10.1038/s41586-023-05736-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-023-05736-y
This article is cited by
-
Characterization of autoimmune eye disease in association with Down’s syndrome
Eye (2024)
-
Safety and Efficacy of Biologic Medications and Janus Kinase Inhibitors in Patients with Down Syndrome: A Retrospective Cohort Study
Clinical Drug Investigation (2024)
-
Symptoms of Catatonia Observed in Down Syndrome Regressive Disorder: A Retrospective Analysis
Journal of Autism and Developmental Disorders (2024)
-
Integration of ATAC-seq and RNA-seq identifies MX1-mediated AP-1 transcriptional regulation as a therapeutic target for Down syndrome
Biological Research (2023)
-
The extrafollicular B cell response is a hallmark of childhood idiopathic nephrotic syndrome
Nature Communications (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
