
Abstract
Alzheimer’s disease (AD) is characterized by deposition of amyloid-β (Aβ) plaques and neurofibrillary tangles in the brain, accompanied by synaptic dysfunction and neurodegeneration. Antibody-based immunotherapy against Aβ to trigger its clearance or mitigate its neurotoxicity has so far been unsuccessful. Here we report the generation of aducanumab, a human monoclonal antibody that selectively targets aggregated Aβ. In a transgenic mouse model of AD, aducanumab is shown to enter the brain, bind parenchymal Aβ, and reduce soluble and insoluble Aβ in a dose-dependent manner. In patients with prodromal or mild AD, one year of monthly intravenous infusions of aducanumab reduces brain Aβ in a dose- and time-dependent manner. This is accompanied by a slowing of clinical decline measured by Clinical Dementia Rating—Sum of Boxes and Mini Mental State Examination scores. The main safety and tolerability findings are amyloid-related imaging abnormalities. These results justify further development of aducanumab for the treatment of AD. Should the slowing of clinical decline be confirmed in ongoing phase 3 clinical trials, it would provide compelling support for the amyloid hypothesis.
Similar content being viewed by others

Main
The amyloid hypothesis posits that Aβ-related toxicity is the primary cause of synaptic dysfunction and subsequent neurodegeneration that underlies the progression characteristic of AD1,2. Genetic, neuropathological, and cell biological evidence strongly suggest that targeting Aβ could be beneficial for patients with AD3,4. So far, attempts at therapeutically targeting Aβ have not been successful5,6,7, casting doubt on the validity of the amyloid hypothesis. However, the lack of success may have been due to the inability of the antibodies to adequately engage their target or the proper target in the brain, or selecting the wrong patient population.
We describe the development of an antibody-based immunotherapeutic approach by selecting human B-cell clones triggered by neo-epitopes present in pathological Aβ aggregates. The screening of libraries of human memory B cells for reactivity against aggregated Aβ led to molecular cloning, sequencing, and recombinant expression of aducanumab (BIIB037), a human monoclonal antibody that selectively reacts with Aβ aggregates, including soluble oligomers and insoluble fibrils. In preclinical studies, we show that an analogue of aducanumab is capable of crossing the blood–brain barrier, engaging its target, and clearing Aβ from plaque-bearing transgenic mouse brains. These results prompted the start of clinical trials8.
We report here interim results from a double-blind, placebo-controlled phase 1b randomized trial (PRIME; ClinicalTrials.gov identifier NCT01677572) designed to investigate the safety, tolerability, pharmacokinetics, and pharmacodynamics of monthly infusions of aducanumab in patients with prodromal or mild AD with brain Aβ pathology confirmed by molecular positron emission tomography (PET) imaging. Together, our data support further development of aducanumab as an Aβ-removing, disease-modifying therapy for AD.
Removal of brain Aβ plaques in patients with AD
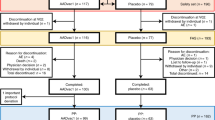
In the PRIME study, 165 patients were randomized and treated between October 2012 and January 2014 at 33 sites in the United States. Patients with a clinical diagnosis of prodromal or mild AD and visually positive Aβ PET scan9 were given monthly intravenous infusions of placebo or aducanumab at doses of 1, 3, 6 or 10 mg kg−1 for one year. Of these patients, 125 completed and 40 discontinued treatment, most commonly due to adverse events (20 patients) and withdrawal of consent (14 patients): 25% of the placebo group discontinued compared with 23%, 19%, 17%, and 38% of the 1, 3, 6 and 10 mg kg−1 aducanumab dose groups, respectively (Extended Data Fig. 1). Baseline characteristics, including cognitive measures, were generally well-balanced across the groups, although the 1 mg kg−1 dose group included a higher proportion of patients with mild AD, and the aducanumab treatment groups tended to have a higher Clinical Dementia Rating—Sum of Boxes (CDR-SB) score (Table 1).
Treatment with aducanumab reduced brain Aβ plaques as measured by florbetapir PET imaging in a dose- and time-dependent fashion (Fig. 1, 2a). The mean PET standard uptake value ratio (SUVR) composite score at baseline was 1.44. After 54 weeks of treatment, this had decreased significantly (P < 0.001) in the 3, 6 and 10 mg kg−1 dose groups; whereas change for the placebo group was minimal (Fig. 2a, Extended Data Table 1). In the 10 mg kg−1 dose group, the SUVR composite score was 1.16 after 54 weeks of treatment, a value near the purported quantitative cut-point of 1.10 that discriminates between positive and negative scans (Fig. 2b)10. The adjusted mean changes in SUVR composite scores in the 6 and 10 mg kg−1 groups treated for 26 weeks were similar in magnitude to the dose group below (3 and 6 mg kg−1, respectively) treated for 54 weeks (Fig. 2a). Reductions in amyloid PET SUVR composite score in aducanumab-treated patients were similar in patients with mild and prodromal AD, and apolipoprotein E (ApoE) ε4 carriers and non-carriers (Extended Data Fig. 2a, b). Pre-specified regional analyses of SUVR changes demonstrated statistically significant dose-dependent reductions in all brain regions, except for the pons and sub-cortical white matter, two areas in which Aβ plaques are not expected to accumulate (Extended Data Fig. 3).

Individuals were chosen based on visual impression and SUVR change relative to average one-year response for each treatment group (n = 40, 32, 30 and 32, respectively). Axial slice shows anatomical regions in posterior brain putatively related to AD pathology. SUVR, standard uptake value ratio.

a–c, Change from baseline (a, analyses using ANCOVA), SUVR values (b), and categorization of change in amyloid PET (c) at week 54 and associated change from baseline CDR-SB and MMSE in aducanumab-treated patients (post hoc analysis). Categorization of amyloid PET at week 54 based on s.d. of change from baseline in placebo-treated patients. **P < 0.01; ***P < 0.001 versus placebo; two-sided tests with no adjustments for multiple comparisons. Mean ± s.e. ANCOVA, analysis of covariance; CDR-SB, Clinical Dementia Rating—Sum of Boxes; MMSE, Mini Mental State Examination; SUVR, standard uptake value ratio.
Effect on clinical measures
Clinical assessments were exploratory as the study was not powered to detect clinical change. The test of dose response was the pre-specified primary analysis for the clinical assessments. Analysis of change from baseline on the CDR-SB (adjusted for baseline CDR-SB and ApoE ε4 status) demonstrated dose-dependent slowing of clinical progression with aducanumab treatment at one year (dose-response, P < 0.05), with the greatest slowing for 10 mg kg−1 (P < 0.05 versus placebo) (Fig. 3a, Extended Data Table 1). Sensitivity analysis using a mixed model for repeated measures (MMRM) also showed a trend for slowing of decline on the CDR-SB at one year (P = 0.07 with 10 mg kg−1 aducanumab versus placebo). A dose-dependent slowing of clinical progression on the Mini Mental State Examination (MMSE) with aducanumab treatment was also observed at one year (dose-response, P < 0.05), with the greatest effects at 3 and 10 mg kg−1 aducanumab (P < 0.05 versus placebo) (Fig. 3b, Extended Data Table 1). On sensitivity analysis using MMRM, the greatest difference was retained for 10 mg kg−1 aducanumab (P < 0.05 versus placebo), with a smaller difference at 3 mg kg−1 (P = 0.10 versus placebo). No changes from baseline after one year were found on the composite neuropsychological test battery (NTB) or the Free and Cued Selective Reminding Test (FCSRT) free recall (Extended Data Table 1), but skewed non-normal (floor) effects at baseline were observed. The floor effects on the NTB were seen in the individual tests; specifically, in the two most clinically relevant components given the stage of the population enrolled: Wechsler Memory Scale-Fourth Edition Verbal Paired Associates II (WMS-IV VPA II) and Rey Auditory Verbal Learning Test (RAVLT) delayed recall of the NTB memory domain.

a, b, Aducanumab effect on CDR-SB (a) and MMSE (b). *P < 0.05 versus placebo; two-sided tests with no adjustments for multiple comparisons. CDR-SB and MMSE were exploratory endpoints. Adjusted mean ± s.e. Analyses using ANCOVA. CDR-SB, Clinical Dementia Rating—Sum of Boxes; MMSE, Mini Mental State Examination.
Safety and tolerability
The most common adverse effects were amyloid-related imaging abnormalities (ARIA), headache, urinary tract infection, and upper respiratory tract infection (Table 2). Using the most specific description of ARIA by magnetic resonance imaging (MRI), ARIA-vasogenic oedema (ARIA-E) abnormalities occurred in no patients receiving placebo compared with 1 (3%), 2 (6%), 11 (37%), and 13 (41%) patients receiving 1, 3, 6 and 10 mg kg−1 aducanumab, respectively (Extended Data Table 2). ARIA-E was generally observed early in the course of treatment, MRI findings typically resolved within 4–12 weeks, and of the 27 patients who developed ARIA-E, 15 (56%) continued treatment (Supplementary Information). All cases of symptomatic ARIA were required to be reported as medically important serious adverse effects. No patients were hospitalised for ARIA. The only serious adverse effects (by preferred term) that occurred in more than one patient in any treatment group were ARIA (0, 1 (3%), 1 (3%), 4 (13%), and 5 (16%) of patients receiving placebo, and 1, 3, 6 and 10 mg kg−1 aducanumab, respectively) and superficial siderosis of the central nervous system (0, 1 (3%), 0, 2 (7%), and 3 (9%) of patients receiving placebo and 1, 3, 6 and 10 mg kg−1 aducanumab, respectively). Owing to the requirement for repeated MRI assessments of those patients who developed ARIA, these individuals were partially unblinded to treatment. Other adverse effects and serious adverse effects were consistent with the patient population. There were no drug-related deaths (Supplementary Information).
Pharmacokinetics
The pharmacokinetics of aducanumab (maximum concentration (Cmax) and cumulative area under the concentration curve (AUC)) were linear across the dose range in patients who received all 14 planned doses (Extended Data Table 3). The median plasma half-life was 21 days. In total, 3 of 118 evaluable patients (3%) in the combined aducanumab groups developed treatment-emergent anti-aducanumab antibodies within the first year of treatment. Antibody responses were transient, with minimal titres, and had no apparent effect on aducanumab pharmacokinetics or safety.
Brain penetration and binding to Aβ plaques
In the preclinical studies which preceded PRIME, systemically administered aducanumab (single dose, 30 mg kg−1 intraperitoneally (i.p.)) bound to diffuse and compact Aβ plaques in the brains of 22-month-old female Tg2576 transgenic mice (‘Target engagement study’; Extended Data Fig. 4a–d). Cmax in plasma was 181 μg ml−1, with a terminal elimination half-life (t1/2) of 2.5 days. The Cmax in brain was 1,062 ng g−1 of tissue, and approximately 400–500 ng g−1 of drug was measured 3 weeks after dosing, suggesting long-term retention. Consequently, the brain:plasma AUC ratio of 1.3% was higher than the 0.1% frequently reported for systemically administered antibodies11,12.
Administration of a single dose of aducanumab did not affect plasma (Extended Data Fig. 4b) or brain (data not shown) Aβ concentrations, consistent with the observation that aducanumab does not bind to soluble Aβ monomers. In contrast, the murine bapineuzumab precursor antibody 3D6, which binds to Aβ monomers, triggered a transient plasma Aβ spike (Extended Data Fig. 4b). Similarly, plasma Aβ concentrations were stable after repeated dosing with aducanumab in the PRIME study (data not shown). Within 24 h of dosing, aducanumab bound to parenchymal brain Aβ with a spatial pattern essentially superimposable with ex vivo pan-Aβ antibody staining, confirming that aducanumab binds all morphological types of brain Aβ plaques in vivo, including diffuse Aβ deposits and compact Aβ plaques (Extended Data Fig. 4c, d). Aducanumab binding to Aβ deposited in cerebral amyloid angiopathy (CAA) lesions within brain blood vessel walls was less prominent than parenchymal Aβ binding, when compared with the total amount of Aβ (Extended Data Fig. 4c, d).
Reduction of brain Aβ in transgenic mice
Exposure in plasma and brain correlated linearly with dose after chronic dosing in plaque-bearing transgenic mice (Extended Data Fig. 5) (Supplementary Information). chaducanumab, a murine IgG2a/κ chimaeric analogue, dose-dependently reduced Aβ measured in brain homogenates by up to 50% relative to the vehicle control in the diethylamine (DEA) fraction that extracted soluble monomeric and oligomeric forms of Aβ40 and Aβ42, and in the guanidine hydrochloride (GuHCl) fraction that extracted insoluble Aβ fibrils (Fig. 4a, b).

a, b, Aβ40 and Aβ42 levels in soluble DEA (a) and insoluble GuHCl (b) brain fractions. c, d, Total brain Aβ (6E10) and compact amyloid plaques (ThioS) in cortex (c) and hippocampus (d) (mean ± s.e.; n = 20–55; dotted line 50% reduction; *P < 0.05 versus control). e–h, ThioS staining of amyloid deposits (e) and Visiopharm software (f) differentiated parenchymal deposits (green) from vascular deposits (red) (representative pictures 10× magnification), and quantified area of vascular amyloid (g, h; mean ± s.e.; n = 20–24).
Quantitative 6E10 immunohistochemistry showed significant reductions in all forms of Aβ deposits by up to 70% (Fig. 4c, d). Thioflavin S (ThioS) staining of compact Aβ plaques showed dose-dependent and statistically significant reductions in the cortex and hippocampus by up to 63% (Fig. 4c, d). Quantitative histology indicated that chaducanumab significantly reduced the number of plaques of all sizes, including plaques >500 μm2 and plaques <125 μm2 (Extended Data Fig. 6a–c). Quantification of ThioS-positive vascular and parenchymal Aβ plaques separately showed that chaducanumab did not affect vascular Aβ in either cortex or hippocampus (Fig. 4e–h).
To identify the mechanism of Aβ clearance, we analysed the involvement of microglia which are known to display enhanced phagocytic activities through binding to the Fc region of an antibody13,14. chaducanumab significantly increased recruitment of Iba-1-positive microglia to Aβ plaques, suggesting FcγR-mediated phagocytosis of antibody–Aβ complexes as a possible clearance mechanism (Extended Data Fig. 7a–c and Supplementary Information).
Biochemical characterization
The apparent affinities of aducanumab and chaducanumab for aggregated Aβ42, with half maximal effective concentration (EC50) values of 0.1 nM, were comparable to 3D6 (ref. 13) (Fig. 5a). Neither aducanumab nor chaducanumab bound monomeric soluble Aβ40 at concentrations up to 1 μM, indicating >10,000-fold selectivity for aggregated Aβ over monomer, whereas 3D6 bound soluble Aβ40 with an EC50 of 1 nM (Fig. 5b). In contrast to 3D6, which immunoprecipitated both monomeric and aggregated Aβ, chaducanumab bound soluble Aβ42 oligomers and insoluble Aβ42 fibrils prepared in vitro, but not Aβ42 monomers (Fig. 5c). Histological staining of autopsy tissue from patients with AD or aged amyloid precursor protein (APP) transgenic mice confirmed binding of aducanumab to bona fide human Aβ fibrils (Fig. 5d, e).

a, Binding of chaducanumab or 3D6 to immobilized fibrillar Aβ42. Mean ± s.d., in triplicate. b, Capture of soluble monomeric Aβ40 with immobilized chaducanumab or 3D6. Mean ± s.d., in triplicate. c, Dot blots of Aβ42 monomer, soluble oligomers, or insoluble fibrils immunoprecipitated with chaducanumab, 3D6, or irrelevant antibody control. Equivalent concentrations confirmed by direct dot blotting (Peptide). d, e, Immunostaining of Aβ in autopsy brain tissue from a patient with AD with chaducanumab (0.2 μg ml−1) (d) and 22-month-old Tg2576 transgenic mouse brain tissue with aducanumab (60 ng ml−1) (e).
Discussion
The PRIME study shows that aducanumab penetrates the brain and decreases Aβ in patients with AD in a time- and dose-dependent manner. Within 54 weeks of treatment, 3, 6 and 10 mg kg−1 doses of aducanumab significantly decreased the amyloid PET SUVR. Patients receiving placebo showed virtually no change in their mean PET SUVR composite scores over one year, indicating that Aβ pathology had already reached an asymptote of accumulation. Considering that it may have taken up to 20 years for Aβ to have accumulated to the levels in these patients at study entry15, the observed kinetics of Aβ removal within a 12-month time period appears encouraging for a disease-modifying treatment for patients with AD.
The cognitive results for CDR-SB and MMSE provide support for the clinical hypothesis that reduction of brain Aβ confers a clinical benefit. Post hoc analysis showed that those aducanumab-treated patients who had decreased SUVR scores >1 standard deviation unit relative to placebo-treated patients after one year of treatment experienced a stabilization of clinical decline on both CDR-SB and MMSE scores; whereas, those patients with a smaller or no decrease experienced clinical decline similar to placebo patients (Fig. 2c). The apparent clinical benefit observed in PRIME could also be explained by the binding of aducanumab to oligomeric forms of Aβ, which would not be directly detected by PET imaging. The reductions in SUVR scores may be surrogates for reductions in toxic soluble Aβ oligomers which may have had a more functionally relevant impact on cognition. Whereas significant Aβ reduction was detectable by 6 months, clinical effects were not apparent until one year. Given that clearance of Aβ could be followed by recovery of neuronal function, a lag between reduction of Aβ burden and slowing of disease progression is not altogether surprising.
The main safety finding, ARIA-E, was dose-dependent and more common in ApoE ε4 carriers, consistent with findings with other anti-Aβ monoclonal antibodies7,16,17. Although the underlying cause of ARIA is not well understood, it is likely that the MRI signal of ARIA is due to increased extracellular fluid. This may be a result of underlying CAA, changes in perivascular clearance and vascular integrity, or local inflammatory processes associated with Aβ-targeting therapies17,18,19,20 (see Supplementary Information for further discussion).
Study limitations of the PRIME phase 1b study included staggered parallel-group design, small sample sizes, limited region (USA only), and possible partial unblinding due to ARIA-E. Measures were taken to maintain blinding to adverse effects: raters of given tests were not permitted to perform other clinical assessments, and were blinded to other assessments (for example, MMSE and CDR raters were required to be different and neither were permitted to perform other study assessments). Post hoc analyses of change from baseline PET SUVR composite score and cognition by presence/absence of ARIA suggested no apparent difference in treatment effect when comparing patients with and without ARIA-E (Extended Data Table 4). There was overlap in enrolment in Arms 1–3 (aducanumab 1 and 3 mg kg−1, placebo) and Arms 4 and 5 (aducanumab 10 mg kg−1, placebo) but Arms 6 and 7 (aducanumab 6 mg kg−1, placebo) were initiated after enrolment in Arms 1–5 was complete. This was a small study designed for assessment of safety and tolerability, and for detecting a pharmacological effect on brain Aβ levels measured by PET imaging. The trial was not powered for the exploratory clinical endpoints, thus the clinical cognitive results should be interpreted with caution. Primary analyses were based on observed data with no imputation for missing values, nominal P values were presented with no adjustments for multiple comparisons, and they were supported by sensitivity analyses using a MMRM.
The initiation of the PRIME study and its results are supported by extensive preclinical data. Detection on parenchymal Aβ plaques following a single systemic administration confirmed that aducanumab penetrates the brain to a sufficient extent to allow accumulation on Aβ plaques. This is consistent with earlier findings showing that, in the presence of significant Aβ deposition, plaque-binding antibodies can be detected bound to the target over an extended period14,21. The minimal effective dose upon repeated systemic administration of chaducanumab in transgenic mice was 3 mg kg−1 (corresponding to minimally effective concentrations of 13.8 ± 1.9 μg ml−1 in plasma and 99.8 ± 30.0 ng g−1 in brain) with reductions of Aβ42 in soluble and insoluble brain fractions of approximately 50%, and reductions in Aβ plaque of approximately 40%. Since exposure at 3 mg kg−1 in animals and humans is approximately equivalent, the observed dose-response in the model was consistent with the clinical doses that led to reductions in amyloid PET SUVR. chaducanumab cleared plaques of all sizes, suggesting that aducanumab triggered clearance of pre-existing Aβ plaques and prevented formation of new plaques.
In transgenic mice, aducanumab preferentially bound to parenchymal Aβ over vascular Aβ deposits, consistent with the lack of effect on vascular Aβ following chronic dosing. The effect of anti-Aβ antibody therapies on the vascular Aβ compartment could be related to micro-haemorrhages or oedema in transgenic mice, and may relate to ARIA in clinical trials22. Nevertheless, the preferential binding of aducanumab to parenchymal versus vascular Aβ may have been critical in allowing the use of relatively high doses in the clinical study so as to achieve robust target engagement in the brains of patients with AD.
Several mechanisms may be involved in aducanumab’s Aβ-lowering activity. The clearance of Aβ deposits was accompanied by enhanced recruitment of microglia. Together with the reduced potency of the aglycosylated form of chaducanumab (data not shown), and the ex vivo phagocytosis data, this suggests that FcγR-mediated microglial recruitment and phagocytosis played an important role in Aβ clearance in these models. Activated microglia appeared to encapsulate the remaining central dense core of plaques in treated animals, possibly isolating them from the surrounding neuropil. It is commonly thought that soluble Aβ oligomers, rather than monomers or plaques, may be the primary toxic species23,24. Considering that Aβ plaques might be a source of Aβ oligomers25,26,27,28, this suggests that treatment with aducanumab might slow their release into the neuropil, thereby limiting their toxic effect on neurons29. In fact, chronic dosing of 18-month-old Tg2576 transgenic mice with chaducanumab led to normalization of neuritic calcium overload in the brain30. Other studies have linked calcium dyshomeostasis in neurons and microglia to binding of Aβ oligomers to metabotropic receptors31,32,33. Aducanumab binding to soluble Aβ oligomers may prevent their interaction with those receptors, thereby preventing the detrimental effect of membrane depolarization. Restoration of this functional endpoint suggests that aducanumab treatment may lead to beneficial effects on neuronal network function underlying cognitive deficits.
Together, the clinical and preclinical data support continued development of aducanumab as a disease-modifying treatment for AD. The clinical study results provide robust support to the biological hypothesis that treatment with aducanumab reduces brain Aβ plaques and, more importantly, to the clinical hypothesis that Aβ plaque reduction confers clinical benefit. This concurs with preclinical data demonstrating brain penetration, target engagement, and dose-dependent clearance of Aβ plaques in transgenic mice. The clinical effects of aducanumab need to be confirmed in larger studies. Both the long-term extension (LTE) phase of this study and phase 3 development are ongoing.
Methods
Clinical study subjects
Patients were screened for inclusion in three stages. First, patients were evaluated on demographic, and clinical and laboratory criteria, including being between 50–90 years of age, and meeting clinical criteria for either prodromal or mild AD, as determined by the investigator. The criteria for prodromal AD were: MMSE score between 24–30 (inclusive), a spontaneous memory complaint, objective memory loss defined as a free recall score of ≤27 on the FCSRT34, a global CDR score of 0.5, absence of significant levels of impairment in other cognitive domains and essentially preserved activities of daily living, and an absence of dementia35. The criteria for mild AD were: MMSE score between 20–26 (inclusive), a global CDR of 0.5 or 1.0, and meeting the National Institute on Aging–Alzheimer’s Association core clinical criteria for probable AD36. Second, patients who remained eligible underwent MRI to exclude those with confounding pathology, including acute or sub-acute micro- or macro-haemorrhage, prior macro-haemorrhage, >4 micro-haemorrhages, superficial siderosis or any finding that might be a contributing cause of the patient’s dementia, pose a risk to the patient, or prevent a satisfactory MRI assessment for safety monitoring. Third, remaining eligible patients underwent a florbetapir PET scan, and those with a positive scan based on a visual assessment, as determined by a qualified reader, were eligible. The Aβ PET screening process has been described in a separate publication9. Stable use of most concomitant background medications was permitted and, in the case of cholinesterase inhibitors and/or memantine, patients were required to be on a stable dose for a minimum of 4 weeks before screening with no adjustment of dosing during the double-blind phase of the study. Patients were excluded if they had a medical condition that might be a contributing cause of cognitive impairment.
Clinical study design
This was a multicentre, randomized, 12-month, double-blind, placebo-controlled, multiple-dose study of aducanumab followed by a 42-month, dose-blinded LTE study in patients with either prodromal or mild AD who were Aβ PET-positive (ClinicalTrials.gov identifier NCT01677572). The primary objective was to evaluate the safety and tolerability of multiple doses of aducanumab in patients with prodromal AD or mild AD dementia. The secondary objectives were to: (i) assess the effect on cerebral Aβ plaque content as measured by 18F-florbetapir PET imaging at week 26; (ii) assess the multiple-dose serum concentrations of aducanumab; and (iii) evaluate the immunogenicity of aducanumab after multiple-dose administration. The key exploratory objectives were assessments of the effect of aducanumab on the following: the clinical progression of AD as measured by change from baseline on the CDR-SB, a NTB, and the FCSRT; disease-related biomarkers in blood, cerebral Aβ plaque content as measured by 18F-florbetapir PET imaging at week 54; and cerebral Aβ plaque content by ApoE ε4 carrier status (carrier/non-carrier). Other exploratory endpoints were change from baseline on the Neuropsychiatric Inventory Questionnaire, Cognitive Drug Research computerized test battery, volumetric MRI, and, in a subset of patients, glucose metabolism as measured by fluorodeoxyglucose PET, functional connectivity by task-free functional MRI, cerebral blood flow by arterial spin labelling MRI, and disease-related biomarkers in cerebrospinal fluid. MMSE was another exploratory assessment.
During the 12-month, double-blind, placebo-controlled phase, patients received aducanumab or placebo by IV infusion once every 4 weeks for 52 weeks. In a staggered, parallel-group design, the treatment arms were enrolled as follows: first Arms 1–3 (aducanumab 1 mg kg−1 (n = 30); aducanumab 3 mg kg−1 (n = 30); placebo (n = 20), respectively) in parallel. Once enrolment was open, Arms 4 and 5 (aducanumab up to 10 mg kg−1 (n = 30) (actual dose 10 mg kg−1); placebo (n = 10), respectively) were enrolled in parallel with Arms 1–3. Once enrolment in Arms 1–5 was complete, enrolment in Arms 6 and 7 (aducanumab up to 30 mg kg−1 (n = 30) (actual dose 6 mg kg−1); placebo (n = 10), respectively) began. The trial was initially designed to dose up to 30 mg kg−1, but when ARIA were detected at 10 mg kg−1 it was decided not to proceed to doses higher than 10 mg kg−1 with repeated infusions. Dose escalation in Arms 4 and 5, and then Arms 6 and 7, was based on review of existing safety, tolerability, and pharmacokinetic data, and recommendation of the external Data Monitoring Committee. Patients were randomized (using a centralized interactive voice and web response System (IXRS)) to a treatment group within Arms 1–3, 4 and 5, or 6 and 7, stratified by ApoE ε4 status (carrier or non-carrier). Patient enrolment was monitored so that the ratio of ApoE ε4 carriers to non-carriers was no more than 2:1 and no less than 1:2. During the overlap in enrolment of Arms 1–3 and Arms 4 and 5, patients were randomized using a minimization algorithm. Patients who discontinued study treatment for any reason were encouraged to remain in the study and complete all assessments during the double-blind period. Patients completing the double-blind period and meeting certain eligibility criteria entered the LTE. After enrolment on Arms 6 and 7 were completed, the protocol was amended to include a titration arm and a corresponding placebo group—Arms 8 and 9. Both the LTE and Arms 8 and 9 are ongoing and were not part of this interim analysis.
Investigators, study site staff (except for a designated pharmacist/technician), and study patients were blinded to the patients’ randomized treatment assignment for the placebo-controlled period. Only the designated pharmacist/technician at each site was aware of the assigned treatment for each patient. Aducanumab was supplied as a sterile clear-to-yellow solution for IV infusion at a dose of 200 mg in 4 ml. For patients randomized to receive aducanumab, undiluted aducanumab (required volume based on patient weight) was added to a 100 ml 0.9% saline bag to reach the assigned dose (an equivalent amount of saline was first withdrawn so that the final total volume of all IV bags was identical). All IV bags (active and placebo (100 ml 0.9% saline)) were covered with a sealed brown light-protective bag to maintain blinding with a label including protocol and patient randomization number.
Cases of ARIA were managed in accordance with protocol-defined rules using centrally read MRI findings coupled with clinical symptoms, if present. The rules were consistent with the guidelines published by the Alzheimer Association Research Roundtable Working Group18. Briefly, patients developing mild ARIA-E or ARIA-H (≤4 incident micro-haemorrhages) without clinical symptoms could continue at the same dose; patients developing moderate or severe ARIA-E without clinical symptoms, or those with ARIA-E accompanied by mild clinical symptoms, could suspend treatment and resume at the next lower dose level once ARIA (and symptoms, if any) resolved. Patients who developed ARIA-E or ARIA-H (≤4 incident micro-haemorrhages) accompanied by moderate, severe, or serious clinical symptoms, >4 incident micro-haemorrhages, any incident macro-haemorrhage, or >1 incident haemosiderosis at any time during the study were to permanently discontinue treatment.
The study was conducted in accordance with the Declaration of Helsinki, and the International Conference on Harmonisation and Good Clinical Practice guidelines, and had ethics committee approval at each participating site. All patients provided written informed consent.
Clinical study assessments
Amyloid plaque content, as measured by florbetapir PET imaging, was assessed at screening, and at weeks 26 and 54. Detailed PET scanning protocols have been described in a separate publication9. Briefly, for each florbetapir scan, a dose of 370 MBq was injected intravenously, with PET scanning starting around 50 min later and continuing for approximately 20 min.
Visual reads, the basis for meeting the inclusion criterion of a positive Aβ PET scan, were based upon PET image data, with the registered MRI and fused PET/MRI data providing supplementary anatomical information. Scans were independently interpreted by two board-certified neuroradiologists who, in accordance with the Amyvid Prescribing Information37, had successfully completed a training programme (provided by the manufacturer using either an in-person tutorial or an electronic process). Images were designated as positive or negative, following guidelines described in the Amyvid Prescribing Information37.
A composite cortical SUVR was computed using a volume-weighted average across six brain regions of interest (frontal, parietal, lateral temporal and sensorimotor, anterior, and posterior cingulate cortices), as previously described16, normalized to whole cerebellar activity10,38.
Clinical tests including the CDR and an NTB (comprising RAVLT Immediate and Delayed Recall, Wechsler Memory Scale Verbal Pair Associate Learning Test Immediate and Delayed Recall, Delis–Kaplan Executive Function System Verbal Fluency Conditions 1 and 2, and the Wechsler Adult Intelligence Scale Fourth Edition Symbol Search and Coding Subsets) were performed during screening and at weeks 26 and 54. The FCSRT was performed at screening and at week 52. These clinical tests were administered by a trained, certified clinician or rater experienced in the assessment of patients with cognitive deficits. When possible, the same rater would administer a given test across all visits. In order to maintain blinding to adverse events, raters were not permitted to perform other clinical assessments, and were blinded to other clinical and safety assessments. The rater who conducted the CDR for a patient could not complete any other rating scales for that same patient, and was blinded to the results of all other cognitive scales.
The following safety assessments were performed at regular intervals: physical examination, neurological examination, vital signs, electrocardiogram, and laboratory safety assessments. During the placebo-controlled period, brain MRI was performed at screening and at weeks 6, 18, 30, 42, and 54, and end of study or termination. The MMSE was completed at screening, and at weeks 24, 52, and end of study or termination, and, in patients who developed ARIA, at every scheduled visit until ARIA resolved.
The concentrations of aducanumab in serum and presence of anti-aducanumab antibodies were determined using validated ELISA techniques (Supplementary Information).
Statistical analysis in the clinical study
This interim analysis included all patients randomized to a fixed-dose regimen and completing the double-blind period of the study (data cut-off February 2015). For all analyses, all patients assigned to placebo were treated as a single group. The safety population was defined as all patients who were randomized and received at least one dose of study treatment. Adverse events were coded using the Medical Dictionary for Regulatory Activities classification. The pharmacodynamic and pharmacokinetic populations were defined as all patients who were randomized, received at least one dose of study treatment, and had at least one post-baseline assessment of the pharmacodynamic parameter or at least one measurable aducanumab concentration in serum, respectively.
The primary analysis of the pharmacodynamic and efficacy data was based on Analysis of Covariance (ANCOVA), adjusting for baseline and ApoE ε4 status (carrier and non-carrier) using observed data. No imputation was performed for missing data. For each time point, adjusted means for each treatment, pairwise adjusted differences with placebo, 95% confidence intervals for the pairwise differences, and associated nominal P values for comparison were calculated. No adjustments were made for multiple comparisons/multiple interim analyses. Dose–response was tested using a linear contrast from the ANCOVA model. The linear contrast test is sensitive to a variety of positive dose–response shapes, including a linear dose–response relationship. This served as the primary analysis for the cognition analyses. To account for missing data, a MMRM was used as a sensitivity analysis for the longitudinal data change from baseline data, adjusting for baseline and ApoE ε4 status (carrier and non-carrier). Visit and treatment group were treated as categorical variables in the model along with their interactions. An unstructured covariance matrix was assumed to model the within-patient variability. This model imposes no assumptions on mean trend and correlation structure, and is considered robust.
Subgroup analyses were performed for change from baseline Aβ PET and change from baseline for cognition measures (CDR-SB and MMSE) for baseline clinical stage and ApoE ε4 status (carrier and non-carrier). The subgroup analysis of the pharmacodynamic and efficacy data was based on ANCOVA, adjusting for baseline and ApoE ε4 status (carrier and non-carrier) (for baseline clinical stage only) using observed data.
Serum pharmacokinetics were determined by nonlinear mixed effects model (NONMEM) approach. Sparse samples in the multiple-ascending-dose study and intensive samples from an earlier single-ascending-dose study8 were combined to construct a population pharmacokinetic model. The model was built in NONMEM software using the first-order conditional estimation with interaction method. Cumulative AUC up to month 12 was estimated for each patient. The plasma terminal elimination half-life was estimated in the pharmacokinetic analysis population. The analysis population for the primary immunogenicity analysis was defined as all patients who were randomized, received study treatment, and had at least one post-dose immunogenicity sample evaluated for immunogenicity.
Interim analyses were specified in the protocol for the purpose of planning future studies; no changes were to be made for this study based on the interim analysis results.
A sample size of 30 patients per treatment group would provide more than 90% power to detect a treatment difference of 1 standard deviation with respect to the reduction of Aβ from baseline, based on comparison of each aducanumab group with placebo, at a two-sided significance level of 0.05, and assuming a dropout rate of 20%.
Transgenic mouse studies
Penetration of aducanumab into the brain and target engagement were assessed in 22-month-old female Tg2576 mice following a single dose of aducanumab at 30 mg kg−1 administered i.p. (‘Target engagement study’; n = 4–5 per time point). The ability of aducanumab to reduce Aβ burden was assessed following chronic treatment of 9-month-old male and female Tg2576 transgenic mice dosed weekly i.p. for 6 months with PBS or 0.3, 1, 3, 10, or 30 mg kg−1 of the murine chimaeric variant chaducanumab (‘Chronic efficacy study’; n = 20–55 per treatment group). An additional dosing study (‘Chronic efficacy study with Agly’; n = 12–14 per treatment group) comparing the plaque clearing ability of chaducanumab to that of an effector function-impaired variant (chaducanumab-Agly) was conducted using a similar study design (chronic treatment of 9.5-month-old Tg2576 transgenic mice dosed weekly i.p. for 6 months with PBS or 3 mg kg−1 of chaducanumab or chaducanumab-Agly).
Mice were killed following anaesthesia with ketamine/xylazine (100/10 mg kg−1 i.p.). Blood was collected by cardiac puncture, and mice were perfused with ice-cold heparinized saline (0.9%) using a peristaltic pump. The brain was removed and halved along the medio-sagittal line. The right hemisphere was frozen on dry ice and stored at −80 °C for biochemical analysis. The left hemisphere was fixed by immersion in 10% neutral buffered formalin.
Size of the treatment groups was determined to take into account natural mortality (10–20%) and high inter-animal variability specific to the Tg2576 strain of mice. No animals were excluded from the analyses, unless the animal died prematurely. ‘n’ reported in the manuscript represents the number of animals in each group that were euthanized as scheduled at the end of the study. The allocation of animals to treatment groups took into account date of birth, gender, and weight at baseline. Each treatment group was balanced for mean age, gender, and mean weight. Dosing solutions were coded with letters so that all experimenters were blinded to the treatment. The labelling of the samples collected did not reflect treatment group, so that experimenters processing and analysing the samples were still blinded. Codes were broken once all analyses were completed, including statistical analysis.
All in-life procedures were conducted in strict accordance with protocols approved by Biogen’s Institutional Animal Care and Use Committee.
Biochemical measurements
Please see Supplementary Information.
Histological assessment
Please see Supplementary Information.
Preparation of different Aβ peptide conformations
Synthetic Aβ1–42 (Aβ42) peptide (AnaSpec, Fremont, California, USA) was reconstituted in hexafluoroisopropanol at a concentration of 1 mg/ml, aliquoted, air-dried, and vacuum-concentrated to form a film, and dissolved in dimethyl sulfoxide (DMSO) at a concentration of 5 mg/ml. Aβ42 oligomers and Aβ42 fibrils were prepared by diluting DMSO-reconstituted monomeric into PBS at a concentration of 100 μg/ml and incubating at 37 °C for at least 3 days and 1 week, respectively. The solution was centrifuged at 14,000g for 15 min at 4 °C, and oligomers were recovered from the supernatant following the shorter incubation, whereas fibrils were recovered from the pellet following the longer incubation. For details on the biophysical characterization of high molecular weight Aβ42 aggregates, please see Supplementary Information.
In immunoprecipitation experiments, samples of freshly prepared monomeric, soluble oligomeric, or insoluble fibrillar Aβ42 were immunoprecipitated with chaducanumab, 3D6 or a murine IgG2a control antibody (P1.17), dot-blotted onto a nitrocellulose membrane, and detected with biotinylated pan-Aβ antibody 6E10. Similar results were observed for chaducanumab when immunoblotted with 3D6.
ELISA
Please see Supplementary Information.
Antibody generation using reverse translational medicine
Aducanumab was derived from a de-identified blood lymphocyte library collected from healthy elderly subjects with no signs of cognitive impairment and cognitively impaired elderly subjects with unusually slow cognitive decline. Memory B cells, isolated from peripheral blood lymphocyte preparations by anti-CD22-mediated sorting were cultured on gamma-irradiated human peripheral blood mononuclear cell feeder layers. Supernatants from isolated B cells were screened for their ability to stain Aβ plaques on brain tissue sections, from either patients with AD or aged APP transgenic mice39, and for their binding to aggregated forms of Aβ40 and Aβ42 in vitro. Positive hits meeting the above criteria were counter-screened to exclude clones cross-reacting with full-length APP expressed on stably transfected HEK293 cells (provided by U. Konietzko, University of Zurich, Switzerland; tested negative for mycoplasma contamination; not independently authenticated). Selected Aβ-reactive B-cell clones were subjected to cDNA cloning of IgG heavy and κ or λ light chain variable region sequences, and sub-cloned in expression constructs using Ig-framework specific primers for human variable heavy and light chain families in combination with human J-H segment-specific primers. Aducanumab was engineered to incorporate glycosylated human IgG1 heavy and human κ light chain constant domain sequences. A murine chimaeric IgG2a/κ version of aducanumab (chaducanumab) was generated for use in chronic efficacy studies in APP transgenic mice. An aglycosylated variant of chaducanumab (chaducanumab-Agly), incorporating a single point mutation (N297Q, using standard Kabat EU numbering) which eliminates N-glycosylation of the Fc region and severely reduces FcγR binding40, was generated to test for Fc-related activities. The recombinant mouse IgG2b monoclonal antibody 3D641 was used as a comparator in some studies.
Ex vivo phagocytosis assay
Please see Supplementary Information.
References
Hardy, J. & Selkoe, D. J. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 297, 353–356 (2002)
Hardy, J. A. & Higgins, G. A. Alzheimer’s disease: the amyloid cascade hypothesis. Science 256, 184–185 (1992)
Ising, C., Stanley, M. & Holtzman, D. M. Current thinking on the mechanistic basis of Alzheimer’s and implications for drug development. Clin. Pharmacol. Ther. 98, 469–471 (2015)
Selkoe, D. J. The therapeutics of Alzheimer’s disease: where we stand and where we are heading. Ann. Neurol. 74, 328–336 (2013)
Cummings, J. L., Morstorf, T. & Zhong, K. Alzheimer’s disease drug-development pipeline: few candidates, frequent failures. Alzheimers Res. Ther. 6, 37 (2014)
Doody, R. S. et al. Phase 3 trials of solanezumab for mild-to-moderate Alzheimer’s disease. N. Engl. J. Med. 370, 311–321 (2014)
Salloway, S. et al. Two phase 3 trials of bapineuzumab in mild-to-moderate Alzheimer’s disease. N. Engl. J. Med. 370, 322–333 (2014)
Ferrero, J. et al. First-in-human, double-blind, placebo-controlled, single-dose escalation study of aducanumab (BIIB037) in mild-to-moderate Alzheimer’s disease. Alzheimers Dement (N Y) (in the press)
Sevigny, J. et al. Amyloid PET screening for enrichment of early-stage Alzheimer disease clinical trials: experience in a phase 1b clinical trial. Alzheimer Dis. Assoc. Disord. 30, 1–7 (2016)
Landau, S. M. et al. Amyloid-β imaging with Pittsburgh compound B and florbetapir: comparing radiotracers and quantification methods. J. Nucl. Med. 54, 70–77 (2013)
Banks, W. A. et al. Passage of amyloid beta protein antibody across the blood-brain barrier in a mouse model of Alzheimer’s disease. Peptides 23, 2223–2226 (2002)
Levites, Y. et al. Insights into the mechanisms of action of anti-Abeta antibodies in Alzheimer’s disease mouse models. FASEB J. 20, 2576–2578 (2006)
Bard, F. et al. Peripherally administered antibodies against amyloid beta-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat. Med. 6, 916–919 (2000)
Bohrmann, B. et al. Gantenerumab: a novel human anti-Aβ antibody demonstrates sustained cerebral amyloid-β binding and elicits cell-mediated removal of human amyloid-β. J. Alzheimers Dis. 28, 49–69 (2012)
Villemagne, V. L. et al. Amyloid β deposition, neurodegeneration, and cognitive decline in sporadic Alzheimer’s disease: a prospective cohort study. Lancet Neurol. 12, 357–367 (2013)
Ostrowitzki, S. et al. Mechanism of amyloid removal in patients with Alzheimer disease treated with gantenerumab. Arch. Neurol. 69, 198–207 (2012)
Sperling, R. et al. Amyloid-related imaging abnormalities in patients with Alzheimer’s disease treated with bapineuzumab: a retrospective analysis. Lancet Neurol. 11, 241–249 (2012)
Sperling, R. A. et al. Amyloid-related imaging abnormalities in amyloid-modifying therapeutic trials: recommendations from the Alzheimer’s Association Research Roundtable Workgroup. Alzheimers Dement. 7, 367–385 (2011)
Barakos, J. et al. MR imaging features of amyloid-related imaging abnormalities. AJNR Am. J. Neuroradiol. 34, 1958–1965 (2013)
Zago, W. et al. Vascular alterations in PDAPP mice after anti-Aβ immunotherapy: Implications for amyloid-related imaging abnormalities. Alzheimers Dement. 9 (Suppl), S105–S115 (2013)
Wang, A., Das, P., Switzer, R. C., III, Golde, T. E. & Jankowsky, J. L. Robust amyloid clearance in a mouse model of Alzheimer’s disease provides novel insights into the mechanism of amyloid-beta immunotherapy. J. Neurosci. 31, 4124–4136 (2011)
Boche, D. et al. Consequence of Abeta immunization on the vasculature of human Alzheimer’s disease brain. Brain 131, 3299–3310 (2008)
Haass, C. & Selkoe, D. J. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer’s amyloid beta-peptide. Nat. Rev. Mol. Cell Biol. 8, 101–112 (2007)
Kayed, R. & Lasagna-Reeves, C. A. Molecular mechanisms of amyloid oligomers toxicity. J. Alzheimers Dis. 33 (Suppl 1), S67–S78 (2013)
Benilova, I., Karran, E. & De Strooper, B. The toxic Aβ oligomer and Alzheimer’s disease: an emperor in need of clothes. Nat. Neurosci. 15, 349–357 (2012)
Koffie, R. M. et al. Oligomeric amyloid beta associates with postsynaptic densities and correlates with excitatory synapse loss near senile plaques. Proc. Natl Acad. Sci. USA 106, 4012–4017 (2009)
Shankar, G. M. et al. Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat. Med. 14, 837–842 (2008)
Condello, C., Yuan, P., Schain, A. & Grutzendler, J. Microglia constitute a barrier that prevents neurotoxic protofibrillar Aβ42 hotspots around plaques. Nat. Commun. 6, 6176 (2015)
Jin, M. et al. Soluble amyloid beta-protein dimers isolated from Alzheimer cortex directly induce tau hyperphosphorylation and neuritic degeneration. Proc. Natl Acad. Sci. USA 108, 5819–5824 (2011)
Kastanenka, K. et al. Amelioration of calcium dyshomeostasis by immunotherapy with BIIB037 in Tg2576 mice. Alzheimers Dement. 9, P508 (2013)
Jarosz-Griffiths, H. H., Noble, E., Rushworth, J. V. & Hooper, N. M. Amyloid-β receptors: the good, the bad, and the prion protein. J. Biol. Chem. 291, 3174–3183 (2016)
Morkuniene, R. et al. Small Aβ1-42 oligomer-induced membrane depolarization of neuronal and microglial cells: role of N-methyl-d-aspartate receptors. J. Neurosci. Res. 93, 475–486 (2015)
Um, J. W. et al. Metabotropic glutamate receptor 5 is a coreceptor for Alzheimer aβ oligomer bound to cellular prion protein. Neuron 79, 887–902 (2013)
Derby, C. A. et al. Screening for predementia AD: time-dependent operating characteristics of episodic memory tests. Neurology 80, 1307–1314 (2013)
Dubois, B. et al. Revising the definition of Alzheimer’s disease: a new lexicon. Lancet Neurol. 9, 1118–1127 (2010)
McKhann, G. M. et al. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 7, 263–269 (2011)
Eli Lilly and Company. Amyvid Prescribing Information. http://www.lilly.com. (2013)
Clark, C. M. et al. Cerebral PET with florbetapir compared with neuropathology at autopsy for detection of neuritic amyloid-β plaques: a prospective cohort study. Lancet Neurol. 11, 669–678 (2012)
Hock, C. et al. Generation of antibodies specific for beta-amyloid by vaccination of patients with Alzheimer disease. Nat. Med. 8, 1270–1275 (2002)
Tao, M. H. & Morrison, S. L. Studies of aglycosylated chimeric mouse-human IgG. Role of carbohydrate in the structure and effector functions mediated by the human IgG constant region. J. Immunol. 143, 2595–2601 (1989)
Johnson-Wood, K. et al. Amyloid precursor protein processing and A beta42 deposition in a transgenic mouse model of Alzheimer disease. Proc. Natl Acad. Sci. USA 94, 1550–1555 (1997)
Acknowledgements
These studies were funded by Biogen. The authors thank the patients and their family members participating in the aducanumab studies, and the PRIME investigators (Supplementary Information) and staff conducting these studies. Medical writing support, under direction of the authors, was provided by A. Smith at Complete Medical Communications, and was funded by Biogen. We thank N. Pederson, J. Dolnikova and E. Garber for help in generating the recombinant antibodies, D. Fahrer, C. Quigley, M. Themeles, X. Zhang and P. Auluck for help in generating the histological data, and K. Mack for editorial support and coordination of the authors in combining the preclinical and clinical work in this manuscript.
Author information
Authors and Affiliations
Contributions
T.B., P.H.W., M.M., T.E., K.R., J.G. and R.M.N. designed the preclinical studies, and J.S., Y.L., J.G., J.F., C.H., R.M.N. and A.S. designed the clinical study. P.C. led the imaging implementation for the clinical study. T.C. and J.O. were clinical study statisticians. T.B., P.H.W., M.M., R.D., F.Q., M.A., M.L., S.C., M.S.B., O.Q.-M., R.H.S., H.M.A., T.E., J.G. and R.M.N. generated, analysed, and/or interpreted data from preclinical studies. T.B., P.H.W., M.M., R.D., F.Q., M.A., M.L., S.C., M.S.B., O.Q.-M., R.H.S., H.M.A., T.E., K.R., J.G., C.H., R.M.N. and A.S. critically reviewed preclinical sections of the manuscript. J.S., P.C., L.W., S.S., T.C., Y.L., J.O., J.F., Y.H., A.M., J.G., C.H., R.M.N. and A.S. analysed and interpreted clinical study data and critically reviewed clinical sections of the manuscript. All authors approved the final version of the manuscript for submission. Biogen and Neurimmune reviewed and provided feedback on the paper. The authors had full editorial control of the paper, and provided their final approval of all content.
Corresponding author
Ethics declarations
Competing interests
J.S., P.C., T.B., P.H.W., L.W., R.D., T.C., Y.L., J.O., F.Q., M.A., M.L., S.C., M.S.B., O.Q.-M., R.H.S., H.M.A., T.E., K.R., J.F., Y.H., A.M. and A.S. are current or former employees and/or shareholders of Biogen. J.S. is an employee of F. Hoffmann-La Roche Ltd., Basel, Switzerland; R.D. is an employee of AbbVie Inc., Worcester, Massachusetts, USA; M.A. is an employee of Substantial Living, San Francisco, California, USA; M.L. is an employee of Novartis, Cambridge, Massachusetts, USA; S.C. is an employee of SynteractHCR, Carlsbad, California, USA; O.Q.-M. is an employee of Shire, Lexington, Massachusetts, USA; R.H.S. and K.R. are employees of Yumanity Therapeutics, Cambridge, Massachusetts, USA; T.E. is an employee of Takeda Pharmaceuticals, Cambridge, Massachusetts, USA; J.F. is retired. M.M., J.G., C.H. and R.M.N. are employees and shareholders of Neurimmune. S.S. was a site investigator for the PRIME study and received consultation fees from Biogen, and has received research support from Functional Neuromodulation, Merck, Genentech, Roche, Lilly, and Avid Radiopharmaceuticals, and consultation fees from Merck, Piramal, Lilly, Genentech, and Roche. He owns no stock options or royalties. Biogen has filed and licensed certain patent applications pertaining to Aducanumab.
Additional information
Reviewer Information Nature thanks L. Lannfelt, R. Thomas and the other anonymous reviewer(s) for their contribution to the peer review of this work.
Extended data figures and tables
Extended Data Figure 1 Participant accounting.
PET, positron emission tomography.
Extended Data Figure 2 Amyloid plaque reduction with aducanumab by baseline clinical stage and baseline ApoE ε4 status.
a, b, Analyses by baseline clinical stage were performed using ANCOVA for change from baseline with factors of: treatment, ApoE ε4 status (carrier and non-carrier) and baseline composite SUVR (a), and for analyses by ApoE ε4 status, using treatment and baseline composite SUVR (b). Adjusted mean ± s.e. ApoE ε4, apolipoprotein E ε4 allele; SUVR, standard uptake value ratio.
Extended Data Figure 3 Amyloid plaque reduction: regional analysis SUVR at week 54.
The boxed area indicates the six regions included in the composite score. *P < 0.05; **P < 0.01; ***P < 0.001 versus placebo; two-sided tests with no adjustments for multiple comparisons. Adjusted mean ± s.e. Analyses using ANCOVA. SUVR, standard uptake value ratio.
Extended Data Figure 4 Brain penetration of aducanumab after a single intraperitoneal administration in 22-month-old Tg2576 transgenic mice.
a, b, Aducanumab levels in plasma and brain (a), and plasma Aβ levels after a single dose (b; n = 4–5; mean ± s.e.). c, d, In vivo binding of aducanumab to amyloid deposits detected using a human IgG-specific secondary antibody (c), and ex vivo immunostaining with a pan-Aβ antibody on consecutive section (d). Examples of a compact Aβ plaque (solid arrow), diffuse Aβ deposit (dashed arrow), and CAA lesion (dotted arrow). CAA, cerebral amyloid angiopathy.
Extended Data Figure 5 Exposure following weekly dosing with chaducanumab in 9.5- to 15.5-month-old Tg2576 transgenic mice.
a, b, chaducanumab concentrations in plasma (a), or DEA-soluble brain extract (b) were measured in samples collected 24 h after the last dose in the ‘Chronic efficacy study’. Mean ± s.e. Dotted lines represent the limits of quantitation of each assay. c, Correlations of drug concentrations in plasma (open circles) or brain (open triangles) with administered dose. The average brain concentrations in the two groups receiving the lowest dose were below the limit of quantitation for that assay, which is indicated by a dotted line on the figure.
Extended Data Figure 6 Treatment with chaducanumab affects plaques of all sizes.
a, Following weekly dosing of chaducanumab in Tg2576 from 9.5–15.5 months of age, amyloid plaques were stained with 6E10 and quantified using Visiopharm software. b, Plaque size was defined by area, and coloured as follows: <125 μm2 (cyan), 125–250 μm2 (green), 250–500 μm2 (pink), and >500 μm2 (red). c, chaducanumab treatment was associated with a significant decrease in plaque number in all size ranges relative to vehicle-treated controls, with reductions of 58%, 68%, 68%, and 53% in the number of plaques for the <125 μm2, 125–250 μm2, 250–500 μm2, and >500 μm2 groups size, respectively. Mean ± s.e.; statistically significant differences from vehicle for each size range are indicated with asterisks; *P < 0.05, Mann–Whitney test.
Extended Data Figure 7 Enhanced recruitment of microglia to amyloid plaques following chaducanumab treatment and engagement of Fcγ receptors.
a, b, Brain sections from either PBS- or chaducanumab-treated mice (‘Chronic efficacy study’; 3 mg kg−1 group) were immunostained for Aβ (6E10; red) and a marker of microglia (Iba1; brown). c, The area of individual amyloid plaques was measured, and Iba1-stained microglia were grouped into two categories, either associated with plaques (within 25 μm of a plaque) or not associated with plaques (>25 μm from a plaque). Plaques with circumferences ≥ 70% surrounded by microglia were quantified and stratified based on plaque size. The fraction of plaques that were at least 70% surrounded by microglia was significantly greater in the chaducanumab-treated group (white bars) compared with the PBS control group (grey bars), for plaques ≥250 μm2. Mean ± s.e.; statistically significant differences from vehicle for each size range are indicated with asterisks; *P < 0.05, Bonferroni’s post hoc test following one-way analysis of variance. All quantifications were done using the Visiopharm software. d, e, FITC-labelled Aβ42 fibrils were incubated with different concentrations of the antibodies before adding to BV-2 microglia cell line (d), or primary microglia (e) for phagocytosis experiment measuring uptake of Aβ42 fibrils into the cells by FACS analysis. Mean ± s.d.
Supplementary information
Supplementary Information
This includes Supplementary Methods, a Supplementary Discussion and Results, and a list of the PRIME investigators. (PDF 249 kb)
Rights and permissions
About this article

Cite this article
Sevigny, J., Chiao, P., Bussière, T. et al. The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature 537, 50–56 (2016). https://doi.org/10.1038/nature19323
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nature19323
This article is cited by
-
A single-domain antibody detects and neutralises toxic Aβ42 oligomers in the Alzheimer’s disease CSF
Alzheimer's Research & Therapy (2024)
-
Protocol for a single-arm, pilot trial of creatine monohydrate supplementation in patients with Alzheimer’s disease
Pilot and Feasibility Studies (2024)
-
Histone deacetylase inhibitors VPA and WT161 ameliorate the pathological features and cognitive impairments of the APP/PS1 Alzheimer’s disease mouse model by regulating the expression of APP secretases
Alzheimer's Research & Therapy (2024)
-
Amyloid-β aggregates activate peripheral monocytes in mild cognitive impairment
Nature Communications (2024)
-
Molecular dynamics simulations to explore the binding mode between the amyloid-β protein precursor (APP) and adaptor protein Mint2
Scientific Reports (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
