
Abstract
Exciting new discoveries have transformed the view of the lysosome from a static organelle dedicated to the disposal and recycling of cellular waste to a highly dynamic structure that mediates the adaptation of cell metabolism to environmental cues. Lysosome-mediated signalling pathways and transcription programmes are able to sense the status of cellular metabolism and control the switch between anabolism and catabolism by regulating lysosomal biogenesis and autophagy. The lysosome also extensively communicates with other cellular structures by exchanging content and information and by establishing membrane contact sites. It is now clear that lysosome positioning is a dynamically regulated process and a crucial determinant of lysosomal function. Finally, growing evidence indicates that the role of lysosomal dysfunction in human diseases goes beyond rare inherited diseases, such as lysosomal storage disorders, to include common neurodegenerative and metabolic diseases, as well as cancer. Together, these discoveries highlight the lysosome as a regulatory hub for cellular and organismal homeostasis, and an attractive therapeutic target for a broad variety of disease conditions.
Similar content being viewed by others

Introduction
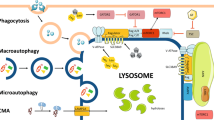
Ever since its discovery by Christian de Duve in the 1950s1, the lysosome has been known as a membrane-enclosed cytoplasmic organelle responsible for the degradation of a variety of biological macromolecules, including proteins, lipids, carbohydrates and nucleic acids2,3. These macromolecules reach the lysosome by various routes, including the endocytic, phagocytic and autophagic pathways, to be degraded in the lysosomal lumen by more than 60 acid hydrolases for subsequent reutilization by the metabolic processes of the cell. In addition to the luminal hydrolases, the lysosome also contains specific sets of integral membrane proteins and lysosome-associated proteins, the latter category being represented by proteins that dynamically interact with the lysosomal surface under certain conditions (Fig. 1).

The lysosome comprises a specific set of luminal, integral-membrane and peripherally associated proteins. The lysosomal lumen contains acid hydrolases that degrade different substrates, enzyme activators and protective factors that aid in the degradation, as well as transport factors such as lipid transfer Niemann–Pick type C2 protein (NPC2), which shuttles free cholesterol to NPC1 for export from the lysosome. The acidic pH of the lysosomal lumen is maintained by a vacuolar ATPase (v-ATPase) embedded in the limiting membrane. In addition, the lysosomal membrane comprises highly glycosylated lysosome-associated membrane proteins (LAMPs) that among other functions protect the lysosomal membrane from degradation, ion channels and transporters that maintain ion homeostasis, cholesterol and other lipid transporters, solute carriers that participate in the export of sugars, nucleosides, amino acids and other products of lysosomal degradation, and SNAREs that mediate fusion of lysosomes with other organelles. The cytosolic face of lysosomes serves as a platform for the dynamic association of proteins and protein complexes (shown in the right part of the figure), including the mechanistic target of rapamycin complex 1 and its regulators that transduce nutrient and growth factor signals (Fig. 2), transcription factors such as TFEB and TFE3 that regulate lysosome biogenesis, autophagy and energy metabolism (Fig. 3), tethering factors that promote lysosome fusion or contacts with other organelles (Figs 4,5), adaptor or scaffold complexes that couple lysosomes to microtubule motors such as kinesins and dynein–dynactin (Fig. 6) and small GTPases that control the recruitment and activation of all of the aforementioned molecules.
Each mammalian cell comprises between 50 and 1,000 lysosomes distributed throughout the cytoplasm. Owing to their role in terminal degradation, lysosomes have often been referred to as the ‘garbage-disposal system’ of the cell. In addition, lysosomes have long been regarded as ‘housekeeping’ organelles that perform their degradative function irrespective of the cell’s status and in relative isolation from other organelles. Moreover, lysosomes have been thought of as static organelles whose cytoplasmic localization does not change over time. Lastly, lysosomal dysfunction has been historically associated with rare diseases such as lysosomal storage disorders (LSDs) caused by impaired degradation of lysosomal substrates. For all of these reasons, the study of lysosomes remained a highly specialized area of research, with few connections to other aspects of cell and organismal physiology.
This narrow view of the lysosome has been dramatically overturned by several recent discoveries. First, lysosomes have been found to participate in many other cellular processes besides degradation, including metabolic signalling, gene regulation, immunity, plasma membrane repair and cell adhesion and migration. Second, the number, composition and functions of lysosomes have been shown to vary in response to environmental cues, as well as to cellular and organismal needs. Third, lysosomes have been found to engage in physical and functional interactions with other cellular structures, including the formation of membrane contact sites. Fourth, lysosomes have been observed to move around the cytoplasm, often changing their size and shape, or undergoing fusion or fission, as they go. Finally, changes in lysosomal function have been implicated in the pathogenesis of common diseases, including neurodegenerative and metabolic disorders, as well as cancer. In this article, we review these recent developments in lysosome biology that now place the lysosome at the centre of a complex regulatory network for the control of cellular and organismal homeostasis.
The lysosome as a signalling hub
Cellular organelles constantly communicate with each other by either establishing contacts or sending signals. While the core function of an organelle is typically executed in the lumen, signalling occurs at its surface. Recent studies revealed that several organelles, including mitochondria4, melanosomes5, peroxisomes6 and lysosomes7, act as ‘launch pads’ for signals to other cellular compartments, including the nucleus8. Here we will focus on specific types of lysosomal signalling pathways that respond to different environmental stimuli and play critical roles in the regulation of various functions, ultimately allowing maintenance of cellular homeostasis.
Lysosomal nutrient sensing and mTORC1 signalling
Being the main mediator of cellular catabolism, the lysosome is in a unique position to use information on cellular degradation and recycling processes as a proxy to sense the cell’s nutritional status. The mechanisms that underlie lysosomal nutrient sensing have recently become a hot topic in cell biology. A major advance in this field was the discovery that nutrient-regulated mechanistic target of rapamycin complex 1 (mTORC1), a key regulator of cellular biosynthetic pathways9, dynamically associates with the lysosome under specific conditions7 (Fig. 2). A key function of mTORC1 is to support cell anabolism and growth in the presence of nutrients and growth factors, while inhibiting catabolic pathways, such as autophagy through the phosphorylation of Unc-51-like kinase 1 (ULK1)10. Of note, mTORC1 also regulates lysosome re-formation during autophagy, a process that helps to restore a full complement of functional lysosomes during prolonged starvation11.

Multiple cellular processes are modulated by signalling pathways initiated from the lysosomal surface. RAG GTPases mediate the dynamic association of several nutrient-modulated protein complexes to the lysosomal surface, including the key regulator of lysosomal function TFEB (Fig. 3), as well as the nutrient sensor and growth regulator mechanistic target of rapamycin complex 1 (mTORC1), together with its regulators tuberous sclerosis complex (TSC), and folliculin (FLCN)–FLCN-interacting protein (FNIP). Ca2+ release from lysosomes modulates the induction of several cellular processes, including lysosomal re-formation from hybrid organelles, endosome–lysosome fusion, TFEB nuclear translocation, autophagosome–lysosome fusion and lysosomal exocytosis. In addition, at least in Caenorhabditis elegans, the lysosomal lipase LIPL-4 generates the lipid oleoylethanolamide (OEA), which binds to lipid-binding protein 8 (LBP8). The OEA–LBP8 complex is exported from the lysosome and translocated into the nucleus for the activation of the nuclear hormone receptors NHR49 and NHR80, which drive genes responsible for the adaptation of mitochondrial metabolism and oxidative stress response, thereby contributing to animal longevity. Lysosomal damage triggers the recruitment of galectins (GAL3, GAL8 and GAL9), which regulate lysosomal removal through autophagy (lysophagy) via the modulation of mTORC1, 5′-AMP-activated protein kinase (AMPK), and Unc-51-like kinase 1 (ULK1)–tripartite motif-containing protein 16 (TRIM16). Small disruptions of the lysosomal membrane activate a lysosomal repair mechanism that is dependent on the recruitment of the endosomal sorting complex required for transport (ESCRT) machinery that allows membrane sealing. The lysosome is also able to sense the incoming autophagic cargo via the recognition of mitochondrial DNA (mtDNA) by Toll-like receptor 9 (TLR9), which is recruited to lysosomes on autophagy induction. This mediates a ‘lysosomal cargo response’ that involves TLR9 activation and consequent recruitment of phosphatidylinositol 4-phosphate 5-kinases (PIP5K) and generation of phosphatidylinositol 4,5-bisphosphate (PtdIns(4,5)P2), which mediates the recycling of SNAREs involved in autophagosome–lysosome fusion. However, to maintain autophagic flux, PtdIns(4,5)P2 generation needs to be controlled in time and space, as PtdIns(4,5)P2 inhibits the lysosomal Ca2+ channel TRPML1, which is required for autophagosome–lysosome fusion events. This control over PtdIns(4,5)P2 levels is provided by the inositol polyphosphate 5-phosphatase activity of Lowe oculocerebrorenal syndrome protein (OCRL). In addition to the regulation of autophagic flux, TLR9-mediated signalling also governs the activation and nuclear translocation of the transcription factor nuclear factor-κB (NF-κB) and an increase in the transcription of proinflammatory cytokine and interferon-β genes, which when uncontrolled can lead to chronic inflammation.
Activation of mTORC1 requires its dynamic recruitment to the lysosomal surface, which is mediated by the amino acid-dependent activation of heterodimeric RAG GTPases and their interaction with Ragulator7,12,13,14,15,16,17. The RAG-dependent recruitment of mTORC1 to the lysosomal surface can also be induced by cholesterol through the involvement of cholesterol-binding Niemann–Pick type C1 protein (NPC1)18. In addition to mTORC1, RAG GTPases modulate the lysosomal recruitment of other nutrient-responsive molecules, including the mTORC1 regulators tuberous sclerosis complex (TSC)19 and folliculin (FLCN)–folliculin-interacting protein 1 (FNIP) complexes20, as well as TFEB21, a master modulator of lysosome biogenesis and autophagy8 (see also the section Lysosomal adaptation) (Fig. 2). Additional details on lysosomal nutrient sensing and on the regulation and functions of mTORC1 at the lysosomal membrane can be found in other recent reviews9,17.
Lysosomal Ca2+ signalling
Lysosomal Ca2+ is key for various lysosomal functions. Release of Ca2+ is required for the fusion of lysosomes with other cellular structures, including endosomes, autophagosomes and the plasma membrane22,23, thereby regulating endocytic membrane trafficking, autophagy and repair of membrane damage (see also the section Interactions with other organelles). Furthermore, lysosomal Ca2+ release is involved in the formation of contact sites with the endoplasmic reticulum (ER), which in turn is able to refill the lysosome with Ca2+ (ref.24). Ca2+ homeostasis is also important for lysosomal acidification, a requirement for the activity of lysosomal hydrolases25. Three main types of Ca2+ channels have been identified in the lysosomal membrane of mammalian cells: transient receptor potential cation channels of the mucolipin family (TRPML), two-pore channels (TPC) and the trimeric Ca2+ two-transmembrane channel P2X4 (refs22,23) Some of these channels are found exclusively on endolysosomes, whereas others have additional locations.
Lysosomal Ca2+ channels respond to a variety of stimuli, such as pH, nutrients and cellular stress, as well as to small molecules such as ATP, nicotinic acid adenine dinucleotide phosphate, phospholipids and sphingosine, suggesting that their activities can be differentially modulated depending on cell conditions, thus allowing more selective Ca2+ signalling responses that are tailored to the needs of the cell.
Perhaps the best characterized lysosomal Ca2+ channel is TRPML1, also known as mucolipin 1 (ref.26). The gene encoding TRPML1 is mutated in an LSD named mucolipidosis type IV, which is characterized by early onset and progressive neurodegeneration27,28. TRPML1 activity mediates Ca2+ release from the lysosomal lumen to the cytosol and can be activated by several stimuli, including starvation29,30 and reactive oxygen species31. TRPML1 is also activated by a specific phosphoinositide, phosphatidylinositol 3,5-bisphosphate, which links lysosomal Ca2+ signalling to intracellular trafficking processes32.
Figure 2 shows the main lysosomal processes that are regulated by TRPML1-mediated Ca2+ release: lysosomal exocytosis and plasma membrane repair33, autophagosome–lysosome fusion26, endosome–lysosome fusion, lysosome size34 and lysosome re-formation from hybrid organelles following fusion35. TRPML1 is also involved in a positive-feedback loop with TFEB, in which TRPML1 regulates TFEB phosphorylation and subcellular localization, while TFEB regulates the expression of the TRPML1 gene29,36 see also the section Lysosomal adaptation) (Figs 2,3). In addition, TRPML1 activity has been associated with specific cellular processes in immune cells, including large particle phagocytosis37, as well as fast and directional migration of dendritic cells through activation of the actin-based motor protein myosin 2 (ref.38). Finally, TRPML1 is a main mediator of the ability of TFEB to promote intracellular clearance of accumulating substrates in LSDs36. Together, these properties make TRPML1 an attractive target for pharmacological modulation in a variety of diseases.

a | TFEB regulates genes involved in several steps of the lysosome–autophagic pathway, including formation of the isolation membrane, conjugation of ubiquitin-like molecules of the Atg8 family (for example, LC3) and elongation of the autophagosomal membrane, cargo recruitment, autophagosome–lysosome fusion, lysosome-mediated degradation and lysosomal biogenesis. For each process, TFEB direct transcriptional targets, identified by chromatin immunoprecipitation–sequencing analyses, are shown in grey boxes (ref.64 and A.B., unpublished observations). b | TFEB nuclear translocation is promoted by multiple environmental stimuli, including starvation, bacterial infections, proinflammatory agents, physical exercise, endoplasmic reticulum (ER) stress, oxidative stress and mitochondrial damage, which allow adaptation of the lysosomal–autophagic pathway to cellular needs. c | TFEB nucleocytoplasmic shuttling is modulated by its phosphorylation status. Activation of the TRPML1 Ca2+ channel and of the phosphatases calcineurin and protein phosphatase 2A (PP2A) results in TFEB dephosphorylation and nuclear translocation29,88. TFEB phosphorylation on serine S142 and S138 residues — which are mechanistic target of rapamycin (mTOR)-dependent phosphorylation sites — promotes its binding to the exportin CRM1 and subsequent nuclear export (ref.97 and A.B., unpublished observations). Whether the nuclear phosphorylation of TFEB is directly or indirectly mediated by mTOR is currently unknown (the former case would necessitate nuclear presence of mTOR). ERK and glycogen synthase kinase 3 (GSK3) have also been proposed as putative kinases for S142/S138 phosphorylation62,98,109. On nuclear export, mechanistic target of rapamycin complex 1 (mTORC1)-mediated phosphorylation of TFEB at S211 induces its binding to 14-3-3 proteins and cytosolic retention92,93,94. v-ATPase, vacuolar ATPase.
Lysosome-dependent cell death and endolysosomal damage response
Several conditions, such as infections and hyperuricaemia, or treatment with lysosomotropic drugs may damage the lysosome by inducing lysosomal membrane permeabilization or rupture. This damage eventually results in leakage of cathepsins, often leading to a form of programmed cell death known as lysosome-dependent cell death39,40,41. Lysosome-dependent cell death can take the form of apoptosis, whereby cathepsins proteolytically activate the proapoptotic proteins BID and BAX, leading to caspase activation39,40, or other forms of cell death such as pyroptosis42, ferroptosis43 and necroptosis44.
Lysosomal damage can also trigger a process known as the endolysosomal damage response45, whereby damaged lysosomes are either eliminated or repaired, to prevent the activation of cell death pathways45 (Fig. 2). Elimination and recycling of damaged lysosomes occurs through a selective autophagy pathway termed ‘lysophagy’46, which is mediated by members of the galectin protein family acting as lysosomal damage sensors. Galectins mediate lysophagy by several mechanisms, including binding of galectin 3 to TRIM16 (ref.47), a member of the tripartite motif (TRIM) protein family48, as well as binding of galectin 8 to the autophagy receptor NDP52 (ref.49). Galectins can also regulate autophagy by both galectin 8-mediated inhibition of mTORC1 and galectin 9-mediated activation of AMPK — a key sensor of reduced cellular energy50. One of the consequences of mTORC1 inhibition caused by lysosomal damage is the activation of TFEB, which induces lysosomal biogenesis and autophagy, further facilitating the recycling of damaged lysosomes and their replacement by newly generated ones45 (see the section Lysosomal adaptation). Recent studies showed that whereas severe lysosomal damage induces galectin-mediated lysophagy, small disruptions instead activate a lysosomal repair mechanism that is independent of lysophagy and is mediated by the Ca2+-dependent recruitment to lysosomal membranes of the endosomal sorting complex required for transport (ESCRT) machinery that seals the damage51,52.
Autophagic cargo sensing and proinflammatory response
Lysosomes sense not only nutrients and metabolites, but also nucleic acids. This task is achieved through members of the Toll-like receptor (TLR) family, which are highly, although not exclusively, expressed in macrophages and dendritic cells53,54. TLRs are able to recognize pathogen-associated molecular patterns, and have a pivotal role in the innate immune response. Of the 13 members of the TLR family, three of them (TLR3, TLR7/8 and TLR9) signal from endolysosomes. These receptors are activated by microbial nucleic acids and, through signal transduction cascades that include different kinds of adaptors, induce the nuclear translocation of either nuclear factor-κB or interferon regulatory factors to stimulate the production of inflammatory cytokines and/or interferons. The endolysosomal site of action of the aforementioned TLRs restricts their function or site of activation to a confined environment; here, the encounter with exogenous nucleic acids is facilitated, because endocytosis is one of the common mechanisms of pathogen entry into host cells55.
TLR9 also has a driving role in the lysosomal response to mitochondrial DNA, which bears similarity to bacterial DNA and can be delivered to the lysosome in the process of autophagy (Fig. 2). This lysosomal cargo response includes local remodelling of phosphoinositides with the production of phosphatidylinositol 4,5-bisphosphate (PtdIns(4,5)P2), followed by the recruitment of the endocytic adaptor AP2 and clathrin, which mediate recycling of SNAREs, such as STX17, used for autophagosome–lysosome fusion. However, PtdIns(4,5)P2 also inhibits the activity of the lysosomal Ca2+ channel TRPML1, which is required for autophagosome–lysosome fusion (Fig. 2). Thus, to sustain the autophagic flux, PtdIns(4,5)P2 levels need to be controlled in time and space. Such control is governed by the inositol polyphosphate 5-phosphatase activity of Lowe oculocerebrorenal syndrome protein (OCRL)56. Overall, this mechanism allows the sensing of the arrival of autophagic cargo at the lysosome to adapt the degradative potential of the cell on autophagy induction.
Apart from supporting autophagic flux, following recognition of mitochondrial DNA, TLR9 also induces nuclear translocation of nuclear factor-κB and subsequent production of proinflammatory cytokines. In steady-state conditions, this is a self-limiting response; however, this response may be sustained under other conditions that impair the degradative capacity of the lysosome, such as LSDs, leading to a pathological inflammation.
Prolongevity lipid signalling from the lysosome
The lysosome has a critical role in the control of lipid homeostasis and in lipid-mediated signalling57. One important lipid family integrated into lysosome biology is the phosphoinositides, which regulate various aspects of lysosome dynamics and function, including its positioning, biogenesis, fusion with autophagosomes and function in lipid transfer at membrane contact sites (recently reviewed elsewhere58). Recent studies performed in Caenorhabditis elegans also revealed a role for the lysosome in prolongevity signalling mediated by the lipid oleoylethanolamide (OEA) and the lipid-binding protein LBP8 (ref.59) (Fig. 2). OEA is generated by lysosomal lypolysis through the activity of lipase LIPL-4, the orthologue of human LIPA. The OEA–LBP8 complex is subsequently exported to the cytosol and translocates to the nucleus to activate the nuclear hormone receptors NHR49 and NHR80 (ref.59). These receptors then induce a transcription programme that regulates mitochondrial β-oxidation and oxidative stress tolerance. This cooperation between lysosomes and mitochondria promotes longevity60. At present, it is unclear whether this pathway also operates to promote longevity in mammals58.
Lysosomal adaptation
Cellular energy metabolism is influenced by environmental cues. Thus, the lysosome should be able to adapt its function in response to diverse environmental conditions to maintain homeostasis. The question that arises from such hypothesis is how the cell modulates the function of an entire organelle, which is made of hundreds of different proteins. An in silico-based approach led to the identification of a transcriptional gene network named ‘CLEAR’ (for ‘coordinated lysosomal expression and regulation’), which includes genes involved in different aspects of lysosomal function and autophagy. This was followed by the discovery of TFEB as the master regulator of this transcriptional network. TFEB and the CLEAR gene network allow lysosomal function and autophagy to be globally controlled61,62.
TFEB and the CLEAR gene network
TFEB belongs to the MiT-TFE family of helix–loop–helix leucine zipper transcription factors together with MITF, TFE3 and TFEC63. TFEB exerts broad control on autophagy and lysosomal function by regulating genes involved in multiple steps of autophagosome biogenesis62, autophagosome–lysosome fusion62,64 and lysosomal degradation pathways61,64, including lysosomal acidification61,64, lysosomal Ca2+ homeostasis36,64, lysosome exocytosis36 and lysosome positioning65 (Fig. 3a). Thus, TFEB behaves as a master regulator of autophagic flux by controlling both cargo delivery and substrate degradation62. Remarkably, TFEB overexpression also increases the number of lysosomes by promoting the biogenesis of new lysosomes. This aspect may have a crucial role in a disease context in which old lysosomes may be dysfunctional due to accumulation of undegraded substrates61,64.
Notably, other members of the MiT-TFE family, such as TFE3, also regulate lysosome biogenesis and autophagy66, and there is evidence for cooperation and partial redundancy between TFEB and TFE3 in the context of metabolic regulation67. Overexpression of TFEB, and in some cases TFE3, promoted intracellular clearance and rescued phenotypic abnormalities in a variety of cellular and mouse models of diseases associated with the accumulation of autophagic/lysosomal substrates, such as several types of LSDs36,68,69, common neurodegenerative diseases70,71,72,73 and metabolic disorders74,75,76. These observations suggest that manipulation of the MiT-TFE-driven CLEAR network may have broad therapeutic applications.
Furthermore, both TFEB and TFE3 regulate other important cellular processes, such as the unfolded protein response77, endocytosis78, stem cell differentiation79,80,81 and endosomal recycling through the retromer complex82. A recent study showed that TFEB and TFE3 also have roles in the regulation of the circadian rhythm, thus linking nutrient-driven and circadian clock-driven regulation of gene expression83. Table 1 lists additional, tissue-specific, functions of TFEB that were identified by in vivo gain-of-function and loss-of-function studies.
Regulation of TFEB by environmental cues
Early studies showed that under normal, steady-state conditions (non-stressed cells under standard culture medium conditions), TFEB is located predominantly in the cytoplasm61, and that its subcellular localization is dependent on nutrient availability, with TFEB translocating to the nucleus on starvation62. A number of conditions were subsequently found to affect TFEB nuclear translocation, including infections84, phagocytosis85, inflammation86, physical exercise87, ER stress77, oxidative stress88 and mitochondrial damage89 (Fig. 3b). The main mechanism that regulates the subcellular localization of TFEB, as well as that of the other MiT-TFE factors, is the phosphorylation status of specific serine residues62,90 (Fig. 3c). The lysosome-associated, nutrient-sensitive mTORC1 kinase has a major role in the phosphorylation of MiT-TFE factors91,92,93,94,95. Notably, TFEB behaves as an atypical mTOR substrate as it binds to RAG GTPases21 and its phosphorylation is resistant to treatment with the mTOR inhibitor rapamycin94.
Whereas mTORC1-mediated phosphorylation inhibits the nuclear translocation of MiT-TFE factors, in a feedback response, MiT-TFE factors promote mTORC1 lysosomal recruitment and activity96. This effect is mediated by the transcriptional induction of RAGD and RAGC to activate mTORC1, thereby enhancing suppression of the CLEAR pathway and favouring anabolism over catabolism96. This mTORC1–TFEB feedback loop has an important role in organismal adaptation to different metabolic conditions, as shown by the impaired ability to reactivate mTOR and protein synthesis in response to leucine after starvation or physical exercise in mice lacking TFEB in liver and muscle, respectively. This metabolic adaptation mechanism is hijacked by cancer cells to support their demanding metabolic needs96 (see also the section Lysosome dysfunction in disease). Overall, this relationship between MiT-TFE factors and mTORC1 links global control of lysosome biogenesis to lysosomal nutrient sensing and explains how lysosomal function can adapt to environmental cues.
In addition to mTOR, other kinases phosphorylate MiT-TFE factors and may affect their subcellular localization, highlighting a complex relationship between MiT-TFE phosphorylation and subcellular localization90. Another important player in the regulation of TFEB nuclear translocation is the Ca2+-dependent phosphatase calcineurin. During starvation, calcineurin is activated by lysosomal Ca2+ release through the Ca2+ channel TRPML1, leading to TFEB dephosphorylation and nuclear translocation29 (Fig. 2). TFEB and TFE3 were also shown to be dephosphorylated by protein phosphatase 2A88.
More recently, it was shown that TFEB continuously shuttles between the cytoplasm and the nucleus, and that its overall localization is the result of the net rates of nuclear import and export97. TFEB nuclear export is mediated by the major exportin CRM1, which binds to a TFEB nuclear export sequence97,98,99. TFEB nuclear export is strictly dependent on the phosphorylation of specific serine residues and is blocked by mTOR inhibition, indicating that mTOR promotes TFEB cytoplasmic localization not only by inhibiting its nuclear import but also by inducing its export97 (Fig. 3c). The picture that emerges from these studies is that of a signalling mechanism94 in which the nucleocytoplasmic shuttling of TFEB, as well as other MiT-TFE transcription factors, conveys information on the lysosomal status to the nucleus.
In addition to phosphorylation-mediated nucleocytoplasmic shuttling, TFEB is also regulated at the level of protein stability. The chaperone-dependent E3 ubiquitin ligase STUB1 is involved in the preferential targeting of phosphorylated TFEB for degradation by the ubiquitin–proteasome pathway100. Finally, a recent study presented evidence of translational regulation of TFEB through spermidine-mediated induction of the translation factor eIF5A. This mechanism was shown to play an important role in the rejuvenation of old human B cells101.
Other transcriptional regulators of lysosome biogenesis
Additional factors were found to regulate lysosomal biogenesis and autophagy. The bromodomain protein BRD4 and the methyltransferase G9a act as repressors of the transcription of lysosomal and autophagy genes102. This pathway appears to be independent of MiT-TFE factors and is induced by starvation through a signalling cascade that involves AMPK and the histone deacetylase SIRT1. The mechanism by which nutrient depletion is relayed to these regulators is still unclear.
STAT3, a member of the STAT family of transcription factors, is also able to regulate lysosomal function. In particular, in the context of mammary gland involution, STAT3 was shown to have a role in the regulation of cathepsins and of lysosome-dependent cell death103. Recent studies showed that STAT3 activity is induced by lysosomal substrate overload in conditions associated with deficiencies of lysosomal proteases, resulting in increased expression of their corresponding genes. This mechanism operates in normally fed cells, and thus is independent of the starvation-induced MiT-TFE pathway104. Independently of its function in the nucleus, STAT3 was also found to enhance the activity of the vacuolar ATPase (v-ATPase) at the lysosome membrane, thereby increasing lysosomal acidification105. This leads to inactivation of STAT3 transcriptional activity and further recruitment of STAT3 to the lysosome. Therefore, STAT3 appears to regulate lysosomal function both through transcription regulation of lysosomal genes in the nucleus and through a transcription-independent regulation of the v-ATPase at the lysosomal membrane105.
Furthermore, AMPK-regulated protein coactivator-associated arginine methyltransferase 1 (CARM1) was shown to act as a transcription coactivator of TFEB in the regulation of lysosomal and autophagic genes106. Finally, the zinc-finger transcription factor ZKSCAN3 and the helix–loop–helix leucine zipper oncoprotein MYC were reported to act as suppressors of lysosomal biogenesis and autophagy by repressing the expression of autophagic and lysosomal genes107,108,109. The MYC inhibitory effect is mediated by competition of MYC with TFEB and TFE3 for the binding to lysosomal and autophagic gene promoters. Inhibition of histone deacetylase activity abolishes MYC promoter binding and allows TFEB and TFE3 to activate lysosomal autophagic gene expression108.
Interactions with other organelles
Although lysosomes were originally thought to exist in relative isolation within the cytoplasm, in recent years they have been found to engage in interactions with other organelles. Some of these interactions lead to fusion, while others involve non-fusogenic contacts with neighbouring organelles (Fig. 4a).

a | Summary of interactions of lysosomes with other organelles (both membrane-bound and membraneless). b | SNARE proteins involved in fusion of lysosomes with other organelles. SNAREs assemble into trans-SNARE complexes comprising a four α-helix bundle at its core. On the basis of the presence of a Q or R residue within the ‘zero’ layer of the bundle, SNAREs are classified as Q-SNAREs (Q) or R-SNAREs (R). c | Machineries involved in fusion of lysosomes with different organelles. Assembly of distinct sets of SNAREs drives fusion of lysosomes with different organelles. Additional factors such as small GTPases (ARL8, RAB7), tethering factors (HOPS, EPG5, PLEKHM1, ATG14L), autophagosome-docking proteins (LC3, GABARAP), regulators (synaptotagmin 7 (SYT7)), BLOC1-related complex (BORC), UVRAG, Rubicon, O-linked N-acetylglucosamine (O-GlcNAc) modification and Ca2+ fluxes, contribute to the specificity, efficiency and regulation of particular fusion events. PtdIns(4,5)P2, phosphatidylinositol 4,5-bisphosphate.
Lysosome fusion with other organelles
All lysosome fusion events — including those with other lysosomes (homotypic fusion) as well as late endosomes, autophagosomes, phagosomes, macropinosomes and the plasma membrane (heterotypic fusion) — are mediated by the assembly of a trans-SNARE complex composed of one R-SNARE and two or three Q-SNAREs (Fig. 4b), and are promoted by the release of Ca2+ from the lumen of the lysosome3 (Fig. 4c). Lysosome fusion with each organelle type depends on a specific trans-SNARE complex and a different set of regulators (Fig. 4b,c). A well-studied fusion event is the merger of lysosomes with late endosomes, the last step in the itinerary of endocytic cargos destined for lysosomal degradation3. Regulators of this particular fusion event include the small GTPase ARL8 and its effector, the heterohexameric tethering complex HOPS, which orchestrate the assembly of the trans-SNARE complex110 (Fig. 4c). ARL8 and another small GTPase, RAB7, additionally interact with the tethering protein PLEKHM1 to promote lysosome–late endosome fusion111,112 (Fig. 4c). Lysosome–lysosome fusion likely relies on the same machinery, except that the R-SNARE VAMP8 replaces VAMP7 in the process113.
A more complex lysosome fusion event is the merger of lysosomes with autophagosomes in the process of autophagy114 (Fig. 4c). Whereas early studies of autophagy focused mainly on the mechanisms of induction and autophagosome formation, in recent years there has been growing interest in the role of the lysosome in autophagic degradation. This role is indeed critical, as lysosomes provide the hydrolases and the acidic pH necessary to degrade the autophagic substrates. Lysosomes also contribute a subset of SNAREs involved in fusion (Fig. 4b,c). Furthermore, lysosomes serve as platforms for various effectors and regulators of autophagosome–lysosome fusion, some of which are similar to those involved in lysosome–late endosome fusion (Fig. 4c); these effectors include the heterooctameric BLOC1-related complex (BORC) that promotes sequential recruitment of ARL8 and HOPS115, and RAB7, which recruits the tethering proteins PLEKHM1 (ref.111) and EPG5 (ref.116), as well as the negative regulator of fusion events, Rubicon117 (Fig. 4c). The lysosome-tethered HOPS, PLEKHM1 and EPG5 in turn interact with phosphatidylethanolamine-conjugated (lipidated) forms of the Atg8/LC3/GABARAP family of docking proteins on the autophagosomal membrane, bringing together the lysosomal and autophagosomal membranes. Lastly, recent studies showed that autophagosome–lysosome fusion is negatively regulated by mTORC1, which catalyses phosphorylation of the tumour suppressor protein UVRAG, leading to its enhanced interaction with Rubicon and decreased interaction with HOPS, and thereby inhibiting fusion events118.
Lysosomes can also fuse with the plasma membrane by a process referred to as ‘lysosomal exocytosis’ (Fig. 4c). This process mediates a number of important lysosomal functions, such as plasma membrane repair33, formation of invasive protrusions in cancer cells119 and secretion of lysosomal contents into the extracellular space36, as happens in the process of bone resorption120.
Maintenance of cellular homeostasis requires that lysosome fusion events are followed by lysosome re-formation from hybrid organelles121. For example, in the process of autophagic lysosome re-formation, prolonged starvation induces the mTORC1-dependent extension of protolysosomal tubules from autolysosomes, which eventually detach and fuse to form new lysosomes11. Likewise, re-formation of lysosomes after their fusion with phagosomes (phagolysosome resolution) also involves extension of protolysosomal tubules122 and regulation by mTORC1 (ref.123). The molecular mechanisms involved in these re-formation processes, as well as in other lysosome tubulation and fission events, are still poorly understood121.
Lysosome contact sites
Over the past few years, it has been realized that lysosomes can also engage in non-fusogenic contacts with other membrane-bound organelles, such as the ER, peroxisomes, Golgi complex and mitochondria (Fig. 4a). These interactions involve the formation of membrane contact sites — organellar domains where closely apposed membranes are held together by various tethering proteins124 (Fig. 5).

Lysosomes establish versatile contacts with various organelles. Several of these contacts now have well-established functional consequences. An important function of contacts with the endoplasmic reticulum (ER) is lipid transfer, likely mediated by STARD3–VAP (step 1), ORP5–NPC1 (step 2), VPS13C–VAP (step 3) and/or RAB7–ORP1L–VAP (step 4) (of note, VAP exists as VAPA and VAPB paralogue with at least partially redundant functions)127,128,129. STARD3, ORP5, Niemann–Pick type C1 protein (NPC1) and ORP1L serve as lipid transfer proteins. VAPs are ER-resident proteins and, other than for their contribution to membrane contacts, their roles in lipid transport processes are not clear. Lysosome–ER contacts also control lysosome positioning and movement. This can occur by the dynamic attachment of lysosomes to perinuclear ER via the RNF26–p62–TOLLIP–USP15 system. Here, lysosomes can be tethered to the ER via p62 in a manner that is regulated by the ubiquitylation status of p62, which is in turn dynamically modulated by the opposing activities of the ubiquitin (Ub) ligase RNF26 and the deubiquitylating enzyme USP15 (step 5)131. Proteins at lysosome–ER contacts also regulate coupling of lysosomes to cytoskeletal motors. For example, ORP1L regulates the cholesterol-dependent coupling of lysosomes to dynein–dynactin via RAB7 and RILP (step 6)130, whereas protrudin regulates the coupling of lysosomes to kinesin 1 via RAB7 and FYCO1 (step 7)154 (Fig. 6). Contacts with the Golgi complex via RAB34, RILP and folliculin promote perinuclear clustering of lysosomes in response to nutrient starvation (step 8)134. Contacts with peroxisomes via synaptotagmin 7 (SYT7) and phosphatidylinositol 4,5-bisphosphate (PtdIns(4,5)P2) mediate NPC1-dependent cholesterol transfer (step 9)133. Annexin 11 (ANXA11)-mediated contacts promote the axonal transport of RNA granules for local translation of mRNAs encoding mitochondrial proteins (step 10)140,141. Finally, lysosomes establish dynamic contacts with mitochondria, where tethering is promoted by RAB7 and counteracted by FIS1, which inhibits RAB7 via the activity of the RAB7 GTPase-activating protein TBC1D15 (step 11)136. For a more detailed description of these mechanisms, see refs3,114,124. PtdIns3P, phosphatidylinositol 3-phosphate; PtdIns4P, phosphatidylinositol 4-phosphate; PtdIns(3,5)P2, phosphatidylinositol 3,5-bisphosphate.
Lysosome–organelle contacts have important functional consequences. For example, maturation of early endosomes to late endosomes and then lysosomes was shown to be accompanied by increased formation of contacts with the ER125. These contacts cause rearrangement of ER tubules125, while also contributing to the budding of recycling endosomal tubules126. Another major function of lysosome–ER membrane contact sites is the non-vesicular transfer of lipids between these organelles. For example, free cholesterol generated by hydrolysis of cholesterol esters in the lumen of lysosomes is exported out of the lysosome by the concerted action of the proteins NPC1 and NPC2 (Fig. 5). Subsequent transfer of free cholesterol into the ER is thought to occur at membrane contact sites and to be mediated by lipid-transfer proteins such as ORP5 and ORP1L, and ER-resident proteins such as VAPA and VAPB127 (Fig. 5). Another lipid-transfer protein, STARD3, mediates cholesterol transport in the opposite direction — from the ER to endolysosomes128. This reverse transport process is independent of the global transcriptional control of cholesterol-regulated genes and might serve to maintain cholesterol homeostasis in endolysosomal organelles without the need for more cholesterol synthesis128. The protein VPS13C has recently been shown to interact with VAPs at lysosome–ER membrane contact sites129 (Fig. 5). VPS13C has an amino-terminal lipid-binding domain that transfers phospholipids between lysosomes and the ER129. The direction and function of this transfer, however, have not been established. In addition to lipid transfer, a number of proteins at lysosome–ER membrane contact sites have been implicated in the regulation of lysosome positioning and movement (see the section Lysosome positioning and motility), in some cases in response to changes in cholesterol or phosphatidylinositol 3-phosphate (PtdIns3P) levels130,131,132 (Fig. 5).
Lysosomes have also been shown to transfer free cholesterol to peroxisomes through membrane contact sites involving lysosomal synaptotagmin 7 and peroxisomal PtdIns(4,5)P2 (ref.133) (Fig. 5). An additional membrane contact site between the lysosome and the Golgi complex contributes to perinuclear clustering of lysosomes and consequent mTORC1 regulation by a non-lysosomal pool of its activator RHEB134,135. This non-lysosomal pool of RHEB is at least in part associated with the Golgi complex itself, and is thought to contribute to mTORC1 activation at lysosome–Golgi contact sites135. Finally, lysosomes form contact sites with mitochondria136. These contacts are promoted by lysosome-associated RAB7 and counteracted by mitochondria-associated FIS1 through recruitment of the RAB7 GTPase-activating protein TBC1D15 (ref.136). In addition to allowing control of lysosomal RAB7 activity by mitochondria, these contacts mark sites for mitochondrial fission, thereby allowing the regulation of both mitochondrial and lysosomal dynamics136 (Fig. 5). Additional molecules have been shown to mediate contacts between the yeast vacuole (the equivalent of mammalian lysosomes) and other organelles137,138,139, but the orthologous molecules have not yet been demonstrated to participate in lysosome contacts in mammalian cells.
Non-fusogenic lysosome interactions are not limited to membrane-bound organelles but have recently been found to include membraneless organelles such as RNA granules140,141 (Fig. 4). RNA granules are condensates of RNAs and proteins formed by liquid–liquid phase separation. Neuronal RNA granules were shown to be tethered by annexin 11 (ANXA11) to a subpopulation of lysosomes141. This tethering allows hitchhiking of the RNA granules on axonal lysosomes towards sites of local protein synthesis, particularly in association with axonal mitochondria and for synthesis of mitochondrial proteins140,141 (Fig. 5).
Lysosome positioning and motility
Another recent development in lysosome biology has been the realization that lysosomes are highly dynamic structures142. Lysosomes are spread throughout the cytoplasm, although with a higher concentration in the perinuclear region. At any given time, some lysosomes are stationary, while others move bidirectionally along microtubule tracks. All lysosomes, however, are potentially motile, as over time some stationary lysosomes start to move, while some moving lysosomes stop moving. Several factors contribute to these dynamics, including immobilization by interactions with perinuclear ER131 and Golgi complex134 (Fig. 5), or peripheral actin filaments underlying the plasma membrane143, as well as mobilization by interaction with microtubule motors144,145 (Fig. 6). Whereas movement from the cell periphery towards the perinuclear region (retrograde movement) is mediated by coupling to cytoplasmic dynein and its activator dynactin145,146, movement from the perinuclear region towards the cell periphery (anterograde movement) is mediated by coupling to several kinesins, including members of the kinesin 1 (KIF5) and kinesin 3 (KIF1) families144,147,148 (Fig. 6).

Several adaptor or scaffold complexes mediate coupling of lysosomes to dynein–dynactin or kinesins (predominantly kinesin 1 (composed of two KIF5 heavy chains and two light chains (KLC), and kinesin 3 (a homodimer of KIF1 heavy chains)) for retrograde and anterograde transport, respectively. These complexes are sensitive to environmental or metabolic conditions, allowing control of lysosome motility and positioning in response to different stimuli. The small GTPases RAB7 and ARL8) regulate lysosome motility and positioning in addition to lysosome interactions with other organelles (Fig. 4). RAB7 regulates coupling of lysosomes to both dynein–dynactin (via RILP)) and kinesin 1 (via FYCO1)). ARL8 regulates coupling of lysosomes to kinesin 1 (via SKIP) and kinesin 3 (probably directly). For a more detailed description of these mechanisms, see refs142,228. BORC, BLOC1-related complex.
Regulation of lysosome positioning
Lysosome positioning and motility are subject to a variety of regulatory inputs. For instance, lysosomes redistribute towards the perinuclear region on removal of amino acids and/or serum (a source of growth factors) from the culture medium149. The mechanisms that govern the perinuclear clustering of lysosomes during starvation are complex, involving effects on the interactions of lysosomes with other organelles and with microtubule motors. As an example of an effect on organelle contacts, serum and amino acid withdrawal induces the association of folliculin with lysosomes, thus initiating a chain of interactions with the dynein–dynactin adaptor RILP and the Golgi complex-localized GTPase RAB34 that ultimately tethers lysosomes to the perinuclear Golgi complex134 (Fig. 5).
Nutrient deprivation also affects the interaction of lysosomes with microtubule motors, both enhancing dynein–dynactin-dependent retrograde transport and inhibiting kinesin-dependent anterograde transport. The enhancement of retrograde transport depends on at least two adaptor systems that independently couple lysosomes to dynein–dynactin: one involving TRPML1 and the Ca2+ sensor ALG2 (ref.150), and the other comprising the lysosomal transmembrane protein TMEM55B and the adaptor protein JIP4 (ref.65) (Fig. 6). Notably, nutrient starvation promotes TFEB/TFE3-regulated transcription of the genes encoding TRPML1 and TMEM55B (refs61,65,66), ensuring long-term enhancement of lysosome retrograde transport. However, because TFEB overexpression has an opposite effect of promoting peripheral redistribution of lysosomes36, TFEB activity may also increase anterograde transport, the overall result being enhanced lysosome dynamics (that is, bidirectional transport between the centre and the periphery of the cell).
The inhibitory effects of starvation on anterograde transport involve other adaptor systems that couple lysosomes to kinesins. One such system consists of BORC and ARL8 (refs148,151,152,153) (Fig. 6). Ragulator physically interacts with BORC, exerting a negative effect on the ability of BORC and ARL8 to recruit kinesins to lysosomes152,153. Starvation strengthens the Ragulator–BORC interaction153, whereas epidermal growth factor stimulation — a potent cue for cell growth — weakens it152, resulting in lysosome redistribution towards the centre or the periphery of the cell, respectively (Fig. 6). An additional system comprises the ER-anchored protein protrudin, which binds to RAB7 and PtdIns3P on the lysosomal membrane to promote the interaction of kinesin 1 with another PtdIns3P-binding adaptor, FYCO1, for movement of lysosomes towards the cell periphery154 (Fig. 6). Amino acid starvation reduces PtdIns3P levels, dissociating protrudin and FYCO1 from lysosomes and thus causing perinuclear clustering of lysosomes132.
Functions influenced by lysosome positioning
In addition to allowing the response to changing nutrient levels, lysosome motility is critical for many other functions, including autophagy, microbial killing, antigen presentation, cell adhesion and migration, and cancer cell invasion155. Several studies showed that lysosome positioning also influences mTORC1 signalling, albeit with variable results. In some studies, peripheral scattering of lysosomes induced by overexpression of ARL8, kinesins, FYCO1 or protrudin enhanced mTORC1 activity, whereas perinuclear clustering of lysosomes caused by knockdown of the same proteins or BORC, or knockout of the corresponding genes, reduced it132,149,156. Since mTORC1 is activated by engagement of growth factor receptors at the plasma membrane, these effects were proposed to depend on the distance of lysosomes from the plasma membrane — the assumption being that the proximity of mTORC1 to the plasma membrane facilitates its activation. Other studies, however, showed that peripheral dispersal of lysosomes by inhibition of dynein157 or acidification of the culture medium158 inhibited mTORC1, whereas perinuclear clustering of lysosomes by human cytomegalovirus infection caused persistent mTORC1 activation159. These effects were attributed to the spatial separation of lysosome-associated mTORC1 and the non-lysosomal pool of RHEB. The discrepancies among these findings may result from the different manipulations that were used to alter lysosome positioning, underscoring the complexity of the mechanisms that link lysosome positioning to mTORC1 signalling.
Lysosome dysfunction in disease
The rapidly expanding knowledge of lysosomal function has resulted in a better understanding of how lysosomal defects can lead to disease. While initially lysosomal dysfunction was identified in rare inherited conditions such as LSDs, more recent studies have uncovered a crucial role of defective lysosomal function in common disease entities such as neurodegenerative diseases, cancer and metabolic disorders. In particular, a decline in lysosomal function with age has been proposed to explain the prevalence of these diseases in elderly individuals160,161.
Lysosomal storage disorders
The paradigmatic example of how dysfunctional lysosomes can cause human disease is represented by LSDs, a group of inherited monogenic diseases characterized by a progressive, multisystemic phenotype often associated with early-onset neurodegeneration. In these diseases, mutations in genes encoding either lysosomal resident proteins or non-lysosomal proteins involved in lysosomal function lead to an impairment of lysosome-mediated degradation and recycling processes, with progressive lysosomal accumulation of undegraded substrates162,163,164,165,166.
LSDs may be caused by mutations in genes encoding lysosomal hydrolases, with consequent primary accumulation of a specific type of undegraded lysosomal substrate. LSDs belonging to this category share extensive similarities in terms of biochemical and cellular mechanisms162,163,164,165,166. A more complex category of LSDs is represented by those caused by mutations in genes that encode lysosomal membrane proteins, which are characterized by broader cellular consequences2. Examples of the latter category include mucolipidosis type IV, which is caused by mutations in the TRPML1 gene and is associated with major intracellular trafficking defects27,28 and Niemann–Pick disease type C1, which is caused by mutations of NPC1 and is characterized by cholesterol accumulation in lysosomes165.
LSDs may also be caused by mutations in genes that encode non-lysosomal proteins that are involved in the sorting, transport and post-translational modification of lysosomal enzymes. For example, mucolipidosis type II is caused by mutations in the gene that encodes N-acetylglucosamine-1-phosphotransferase, which participates in the mannose 6-phosphate-mediated trafficking of lysosomal enzymes162. In multiple sulfatase deficiency, mutations in the sulfatase-modifying factor 1 gene (SUMF1), which encodes an ER-resident protein responsible for the post-translational modification of all sulfatases, result in a severe type of neurodegenerative LSD caused by the deficiency of several lysosomal as well as non-lysosomal sulfatases167,168. Similarly, mutations in the gene that encodes the ER-associated protein CLN8, which is involved in the transport of lysosomal enzymes from the ER to the Golgi complex169, cause a specific type of neuronal lipofuscinosis, an LSD associated with early-onset neurodegeneration and blindness162. Recent studies showed that LSDs may also be caused by mutated subunits of the endolysosomal multisubunit tethering complexes HOPS and CORVET such as VPS33A170,171,172.
Initially, the pathogenesis of LSDs appeared simple and directly correlated to the primary storage of accumulating lysosomal substrates. However, more recent studies based on genomic, cell biology and pathophysiology approaches have identified secondary pathways that have crucial roles in the disease phenotype. For instance, several studies have shown that in most LSDs there is a block of autophagy due to impaired fusion between autophagosomes and lysosomes173,174,175. This block is caused by abnormal cholesterol accumulation in the lysosomal membrane and consequent reduction of the sorting and recycling of SNARE proteins176. As a consequence of a block in autophagy, cells exhibit secondary accumulation of autophagy substrates such as aggregation-prone proteins and dysfunctional mitochondria, leading to inflammation and neurodegeneration, which are late-stage features of LSDs174. Consistent with these observations, autopsy specimens from patients with LSDs showed neuropathological features that are typically detected in patients with common neurodegenerative diseases, such as phosphorylated tau aggregates, neurofibrillary tangles and accumulation of α-synuclein174. These findings demonstrated a mechanistic link between the early-onset neurodegeneration found in LSDs and late-onset neurodegenerative diseases. In addition to neurodegeneration, defective autophagy also appears to be a major factor for LSD skeletal and growth abnormalities through impairment of collagen secretion and reduced bone growth177.
Another important secondary consequence of lysosomal dysfunction in LSDs is the deregulation of mTORC1 activity. Recent studies using cellular and animal models showed that mTORC1 activity is perturbed in several LSDs. However, the nature of this perturbation appears to be cell type specific and disease specific. For example, in Pompe disease myofibers178, as well as in a fly model of Gaucher disease179, mTORC1 activity was reduced. Conversely, in chondrocytes from mouse models of mucopolysaccharidosis types VI and VII and of Niemann–Pick disease type C1, mTORC1 activity was increased177. A possible explanation for these differences may be the nature of the primary and secondary substrates accumulating in different tissues in these LSDs. A notable example is represented by cholesterol, which is known to activate mTORC1 via an SLC38A9–NPC1 lysosomal signalling complex18. Consistently, inhibition of mTORC1 by either rapamycin or starvation significantly ameliorates lysosomal and autophagy defects in cellular models of both mucopolysaccharidosis type VII177 and Niemann–Pick disease type C1 (ref.180).
Neurodegenerative diseases
In addition to LSDs, lysosomal dysfunction has been identified in several common neurodegenerative diseases181, including late-onset forms of neurodegeneration (for example, Parkinson disease, Alzheimer disease and Huntington disease)182, amyotrophic lateral sclerosis183, dementia with Lewy bodies184 and the most common neuromuscular disorder, Charcot–Marie–Tooth disease185,186.
Specific forms of common neurodegenerative diseases may be caused by mutations of genes involved in lysosomal function. Patients with Alzheimer disease who carry mutations in the presenilin 1 gene (PSEN1) show lysosomal and autophagic dysfunction187 owing to defective lysosomal acidification and abnormalities of Ca2+ homeostasis188,189. Likewise, a significant number of patients with Parkinson disease carry mutations in lysosomal–autophagic genes, which represent predisposing factors for disease pathogenesis190,191. Importantly, heterozygosity for mutations in the gene encoding the lysosomal enzyme β-glucocerebrosidase (GBA), whose homozygous mutations cause the LSD Gaucher disease, is a major predisposing factor in Parkinson disease192.
Lysosomal–autophagic dysfunction may also contribute to the pathogenesis of common neurodegenerative diseases caused by protein aggregation. Indeed, in Parkinson disease and related ‘synucleinopathies’193, impairment of the lysosomal–autophagic pathway contributes to α-synuclein accumulation and aggregation within Lewy bodies and neurites of neurons194. In addition, it has been observed that defects in endolysosomal transport result in alterations of amyloid-β peptide production in Alzheimer disease195.
Mounting evidence also suggests that protein aggregation itself may affect the lysosomal–autophagic pathway, thus generating a vicious cycle that boosts protein aggregation and toxicity. For example, in Huntington disease, the aggregation-prone protein huntingtin inhibits autophagosome biogenesis, transport and recognition of expanded huntingtin cargo196,197,198, whereas in Parkinson disease α-synuclein aggregates can lead to lysosomal rupture and consequent cathepsin B-dependent increase in the level of reactive oxygen species199. In addition, α-synuclein toxicity has been associated with a progressive decline of lysosomal function due to cytoplasmic retention of TFEB71. Furthermore, in Down syndrome (trisomy 21), a neurodevelopmental disorder that predisposes to early-onset Alzheimer disease, the extra copy of the gene that encodes amyloid precursor protein (APP) on chromosome 21 leads to increased production of the β-cleaved carboxy-terminal fragment of APP (APP-β-CTF or C99), which impairs lysosomal acidification and function through inhibition of the v-ATPase200. Finally, massive accumulation of lysosome-like organelles was detected at Alzheimer disease amyloid plaques, and in particular in regions where swollen axons contact amyloid deposits. This population of lysosomes had low luminal protease content, suggesting a block of endolysosomal maturation201.
Together, these studies highlight the links between defects in lysosomal–autophagic pathways and the pathogenesis of neurodegeneration, thereby opening up potential therapeutic opportunities for targeting the lysosome to counteract these devastating diseases. In line with this, several studies have shown that in mouse models of neurodegenerative diseases, induction of lysosomal biogenesis and autophagy through viral-mediated overexpression of TFEB results in intracellular clearance of accumulating substrates and significant rescue of the disease phenotype202.
Cancer
For a long time, the lysosome has been considered an attractive therapeutic target for cancer. This was mainly linked to evidence that induction of lysosome-dependent cell death39,203 is an effective therapeutic approach in a variety of cancer types204. However, recent studies indicate that the role of the lysosome in cancer extends beyond its role in cell death and can be linked to its ability to fuel the increased demand of cancer cells for energy sources205. This is particularly important in conditions in which insufficient vascularization of tumours may limit nutrient availability. Several cancer types, such as pancreatic, lung, breast and prostate cancers, as well as glioblastoma and melanoma, have been shown to rely on the induction of lysosomal–autophagic degradation and recycling processes, which act as nutrient-scavenging pathways205.
Considering the role of the lysosomal–autophagic pathway in supporting proliferation and growth of cancer cells, it is not surprising that several types of cancer, including renal cell carcinoma, melanoma, alveolar soft part sarcoma and pancreatic adenocarcinoma, are associated with overexpression of MiT-TFE genes206,207. In these tumours, induction of mTORC1 by MiT-TFE factors (see the earlier discussion in the section Regulation of TFEB by environmental cues’) allows the concomitant hyperactivation of both autophagy-mediated nutrient-scavenging processes and the mTORC1-mediated biosynthetic pathway. Thus, the aberrant activation of both catabolic and biosynthetic pathways supports the energy-demanding metabolism of cancer cells96.
Lysosomes also participate in other processes that promote cancer cell proliferation, invasion and metastasis. An acidic tumour microenvironment, in particular, has been shown to cause redistribution of lysosomes towards the cell periphery208, a process that may enhance proliferation through increased mTORC1 and mTORC2 signalling149,156, as well as invasion and metastasis through exocytosis of lysosomal hydrolases209, matrix metalloproteinases210 and integrins211. A recent study using C. elegans as a model system uncovered a novel mechanism by which lysosome exocytosis promotes cell invasion during normal development119. In this study, interaction of netrin with its receptor on anchor cells was shown to induce lysosome exocytosis leading to the formation of metalloproteinase-enriched protrusions that breach the basement membrane and thus allow the invasion of vulval tissue by the anchor cells119. The orthologue of netrin in humans, netrin 1, is overexpressed in various metastatic cancers, and also stimulates cancer cell invasion212, suggesting that a similar mechanism of lysosomal exocytosis may underlie the spreading of cancer cells. This role has prompted studies to identify pharmacologic agents that interfere with lysosome dispersal towards the cell periphery as a potential cancer therapy213.
Metabolic disorders
Lysosomes have also been implicated in the pathogenesis of various metabolic disorders, as best exemplified by their roles in obesity and diabetes214,215,216. The lipid overload that is characteristic of obesity impairs lysosomal function by various mechanisms214. For instance, feeding of mice with a high-fat diet inhibits lysosomal acidification and acid hydrolase activity, and triggers permeabilization of the lysosomal membrane, with consequent impairment of lysosomal and autophagic functions in different tissues217. Despite the harmful effects of a high-fat diet, lysosomal adaptation via TFEB and TFE3 can exert a protective effect on whole-body lipid and glucose homeostasis, thereby lessening the tendency towards obesity and diabetes67.
Lysosomes also participate in whole-body glucose regulation by maintaining the fitness of insulin-producing pancreatic β-cells215. An early response of these cells to nutrient starvation is the degradation of nascent insulin granules by sequestration into Golgi complex-derived double-membraned structures that subsequently fuse with lysosomes — a process distinct from canonical autophagy218. Lysosomal degradation of nascent insulin granules decreases the secretion of insulin and provides the β-cells with an alternative source of amino acids during fasting. When subjected to a high-fat diet, β-cells induce canonical autophagy, which protects them from ER stress and other harmful effects of lipid oversupply219. However, prolonged lipid overload overwhelms the protective effects of lysosomes and autophagy on metabolic homeostasis and eventually leads to lysosome damage, resulting in deterioration of lysosomal function and β-cell loss.
Conclusions and perspective
The studies reviewed here make it clear that the degradative function of lysosomes is intimately linked to multiple pathways that control cell and organismal homeostasis. Past are the days when lysosomal function could be studied in isolation from the functions of other cellular organelles and processes. Instead, current studies must address the functions of lysosomes in the context of whole cells and organisms, in all of their broad diversity.
Despite the explosion in the understanding of the connection between lysosomes and other cellular structures and pathways, current knowledge of this connection may just represent the tip of the iceberg. Indeed, the lysosomal membrane comprises hundreds of integral and peripheral proteins, many of which are of unknown function. What other nutrient and ion transporters, scaffolds and signalling proteins might connect lysosomal function to cellular metabolism? What other lysosome signalling pathways exist in specialized cell types? In addition to the pathways that were reviewed here (transcriptional regulation by MiT-TFE, BRD4, STAT3 and ZKSCAN3 factors), are there other pathways that mediate global adaptation of lysosomal function to environmental, developmental or pathological cues? Do lysosome-related organelles such as cytotoxic T cell granules, melanosomes and platelet dense bodies share signalling pathways with lysosomes or do they have their own, distinct pathways? Even in cells that do not have specialized lysosome-related organelles, there is evidence for heterogeneity of lysosomes. For example, lysosomes can exhibit different size, shape, positioning, motility, luminal pH, acid hydrolase content and other properties131,150,155,220,221,222,223,224. Do these various lysosome subtypes have different functions? Are there specific populations of lysosomes involved in different cellular functions, for example signalling versus exocytosis? Finally, the ‘life cycle’ of lysosomes remains poorly understood. For example, the understanding of how they fuse and interact with other organelles is still incomplete. Even less well understood is how lysosomes fission and how they re-form after fusing with autophagosomes, phagosomes or other hybrid organelles121. These questions are just examples of all the exciting work that lies ahead to fully elucidate the multiple roles of lysosomes within the cell.
Also promising are the prospects for applying the knowledge gained from fundamental studies of lysosome biology to the treatment of lysosome-related diseases. Considerable advances have already been made in the therapy for LSDs by replacement of the defective lysosomal hydrolases (enzyme replacement) and use of small molecules that inhibit the synthesis of the accumulating substrates (substrate reduction) or promote the folding and stability of mutant lysosomal hydrolases (with the use of chemical chaperones)162,163,164,165. Some LSDs have also been successfully treated by gene therapy to express the normal form of the defective lysosomal hydrolase162,163,164,165. A current challenge is to translate knowledge of lysosome biology to the treatment of more common disorders such as neurodegeneration, cancer and metabolic diseases. In this regard, the use of mTOR inhibitors is being explored to enhance autophagy in neurodegenerative diseases caused by accumulation of protein aggregates225. Lysosomotropic agents such as chloroquine and hydroxychloroquine are being repurposed to inhibit lysosomal degradation and induce lysosome-dependent cell death in cancer cells226. Autophagy inhibitors and lysosomotropic agents may also enhance insulin secretion by pancreatic β-cells in certain diabetic conditions227. As knowledge of the multiple functions of lysosomes in the maintenance of cellular homeostasis expands, we can look forward to the development of better therapeutic interventions for these and other lysosome-related disorders.
References
De Duve, C., Pressman, B. C., R, G. I., Wattieaux, R. & Appelmans, F. Tissue fractionation studies. 6. intracellular distribution patterns of enzymes in rat-liver tissue. Biochem. J. 60, 604–617 (1955).
Saftig, P. & Klumperman, J. Lysosome biogenesis and lysosomal membrane proteins: trafficking meets function. Nat. Rev. Mol. Cell Biol. 10, 623–635 (2009).
Hesketh, G. G., Wartosch, L., Davis, L. J., Bright, N. A. & Luzio, J. P. The lysosome and intracellular signalling. Prog. Mol. Subcell. Biol. 57, 151–180 (2018).
Tait, S. W. & Green, D. R. Mitochondria and cell signalling. J. Cell Sci. 125, 807–815 (2012).
Schiaffino, M. V. et al. Ocular albinism: evidence for a defect in an intracellular signal transduction system. Nat. Genet. 23, 108–112 (1999).
Tripathi, D. N. & Walker, C. L. The peroxisome as a cell signaling organelle. Curr. Opin. Cell Biol. 39, 109–112 (2016).
Sancak, Y. et al. Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell 141, 290–303 (2010). This landmark article demonstrates that mTORC1 exerts its activity on the lysosomal surface to which it is recruited by the nutrient-activated RAG–Ragulator complex.
Settembre, C., Fraldi, A., Medina, D. L. & Ballabio, A. Signals from the lysosome: a control centre for cellular clearance and energy metabolism. Nat. Rev. Mol. Cell Biol. 14, 283–296 (2013).
Saxton, R. A. & Sabatini, D. M. mTOR signaling in growth, metabolism, and disease. Cell 169, 361–371 (2017).
Hosokawa, N. et al. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol. Biol. Cell 20, 1981–1991 (2009).
Yu, L. et al. Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature 465, 942–946 (2010).
Sancak, Y. et al. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science 320, 1496–1501 (2008).
Kim, E., Goraksha-Hicks, P., Li, L., Neufeld, T. P. & Guan, K. L. Regulation of TORC1 by Rag GTPases in nutrient response. Nat. Cell Biol. 10, 935–945 (2008).
de Araujo, M. E. G. et al. Crystal structure of the human lysosomal mTORC1 scaffold complex and its impact on signaling. Science 358, 377–381 (2017).
Lawrence, R. E. et al. A nutrient-induced affinity switch controls mTORC1 activation by its Rag GTPase-Ragulator lysosomal scaffold. Nat. Cell Biol. 20, 1052–1063 (2018).
Su, M. Y. et al. Hybrid structure of the RagA/C-Ragulator mTORC1 activation complex. Mol. Cell 68, 835–846.e3 (2017).
Lawrence, R. E. & Zoncu, R. The lysosome as a cellular centre for signalling, metabolism and quality control. Nat. Cell Biol. 21, 133–142 (2019).
Castellano, B. M. et al. Lysosomal cholesterol activates mTORC1 via an SLC38A9-Niemann-Pick C1 signaling complex. Science 355, 1306–1311 (2017).
Demetriades, C., Doumpas, N. & Teleman, A. A. Regulation of TORC1 in response to amino acid starvation via lysosomal recruitment of TSC2. Cell 156, 786–799 (2014).
Petit, C. S., Roczniak-Ferguson, A. & Ferguson, S. M. Recruitment of folliculin to lysosomes supports the amino acid-dependent activation of Rag GTPases. J. Cell Biol. 202, 1107–1122 (2013).
Martina, J. A. & Puertollano, R. Rag GTPases mediate amino acid-dependent recruitment of TFEB and MITF to lysosomes. J. Cell Biol. 200, 475–491 (2013).
Morgan, A. J., Platt, F. M., Lloyd-Evans, E. & Galione, A. Molecular mechanisms of endolysosomal Ca2+ signalling in health and disease. Biochem. J. 439, 349–374 (2011).
Li, P., Gu, M. & Xu, H. Lysosomal ion channels as decoders of cellular signals. Trends Biochem. Sci. 44, 110–124 (2019).
Wang, W. et al. A voltage-dependent K+ channel in the lysosome is required for refilling lysosomal Ca2+ stores. J. Cell Biol. 216, 1715–1730 (2017).
Mindell, J. A. Lysosomal acidification mechanisms. Annu. Rev. Physiol. 74, 69–86 (2012).
Cheng, X., Shen, D., Samie, M. & Xu, H. Mucolipins: intracellular TRPML1-3 channels. FEBS Lett. 584, 2013–2021 (2010).
Bassi, M. T. et al. Cloning of the gene encoding a novel integral membrane protein, mucolipidin-and identification of the two major founder mutations causing mucolipidosis type IV. Am J. Hum. Genet. 67, 1110–1120 (2000).
Bargal, R. et al. Identification of the gene causing mucolipidosis type IV. Nat. Genet. 26, 118–123 (2000).
Medina, D. L. et al. Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat. Cell Biol. 17, 288–299 (2015).
Wang, W. et al. Up-regulation of lysosomal TRPML1 channels is essential for lysosomal adaptation to nutrient starvation. Proc. Natl Acad. Sci. USA 112, E1373–E1381 (2015).
Zhang, X. et al. MCOLN1 is a ROS sensor in lysosomes that regulates autophagy. Nat. Commun. 7, 12109 (2016).
Dong, X. P. et al. PI(3,5)P2 controls membrane trafficking by direct activation of mucolipin Ca2+ release channels in the endolysosome. Nat. Commun. 1, 38 (2010).
Reddy, A., Caler, E. V. & Andrews, N. W. Plasma membrane repair is mediated by Ca2+ regulated exocytosis of lysosomes. Cell 106, 157–169 (2001).
Cao, Q., Yang, Y., Zhong, X. Z. & Dong, X. P. The lysosomal Ca2+ release channel TRPML1 regulates lysosome size by activating calmodulin. J. Biol. Chem. 292, 8424–8435 (2017).
Miller, A. et al. Mucolipidosis type IV protein TRPML1-dependent lysosome formation. Traffic 16, 284–297 (2015).
Medina, D. L. et al. Transcriptional activation of lysosomal exocytosis promotes cellular clearance. Dev. Cell 21, 421–430 (2011).
Samie, M. et al. A TRP channel in the lysosome regulates large particle phagocytosis via focal exocytosis. Dev. Cell 26, 511–524 (2013).
Bretou, M. et al. Lysosome signaling controls the migration of dendritic cells. Sci Immunol. 2, eaak9573 (2017).
Aits, S. & Jäättelä, M. Lysosomal cell death at a glance. J. Cell Sci. 126, 1905–1912 (2013).
Repnik, U., Stoka, V., Turk, V. & Turk, B. Lysosomes and lysosomal cathepsins in cell death. Biochim. Biophys. Acta 1824, 22–33 (2012).
Wang, F., Gómez-Sintes, R. & Boya, P. Lysosomal membrane permeabilization and cell death. Traffic 19, 918–931 (2018).
Shi, J., Gao, W. & Shao, F. Pyroptosis: gasdermin-mediated programmed necrotic cell death. Trends Biochem. Sci. 42, 245–254 (2017).
Cao, J. Y. & Dixon, S. J. Mechanisms of ferroptosis. Cell Mol. Life Sci. 73, 2195–2209 (2016).
Vanden Berghe, T. et al. Necroptosis, necrosis and secondary necrosis converge on similar cellular disintegration features. Cell Death Differ. 17, 922–930 (2010).
Papadopoulos, C. & Meyer, H. Detection and clearance of damaged lysosomes by the endo-lysosomal damage response and lysophagy. Curr. Biol. 27, R1330–R1341 (2017).
Maejima, I. et al. Autophagy sequesters damaged lysosomes to control lysosomal biogenesis and kidney injury. EMBO J. 32, 2336–2347 (2013).
Chauhan, S. et al. TRIMs and galectins globally cooperate and TRIM16 and galectin-3 co-direct autophagy in endomembrane damage homeostasis. Dev. Cell 39, 13–27 (2016).
Reymond, A. et al. The tripartite motif family identifies cell compartments. EMBO J. 20, 2140–2151 (2001).
Thurston, T. L., Wandel, M. P., von Muhlinen, N., Foeglein, A. & Randow, F. Galectin 8 targets damaged vesicles for autophagy to defend cells against bacterial invasion. Nature 482, 414–418 (2012).
Jia, J. et al. Galectins control mTOR in response to endomembrane damage. Mol. Cell 70, 120–135 (2018).
Radulovic, M. et al. ESCRT-mediated lysosome repair precedes lysophagy and promotes cell survival. EMBO J. 37, e99753 (2018).
Skowyra, M. L., Schlesinger, P. H., Naismith, T. V. & Hanson, P. I. Triggered recruitment of ESCRT machinery promotes endolysosomal repair. Science 360, eaar5078 (2018). This study elegantly shows that the ESCRT machinery is recruited to injured endolysosomes to allow their recovery from damage.
Matz, K. M., Guzman, R. M. & Goodman, A. G. The role of nucleic acid sensing in controlling microbial and autoimmune disorders. Int. Rev. Cell Mol. Biol. 345, 35–136 (2019).
Vidya, M. K. et al. Toll-like receptors: significance, ligands, signaling pathways, and functions in mammals. Int. Rev. Immunol. 37, 20–36 (2018).
Majer, O., Liu, B. & Barton, G. M. Nucleic acid-sensing TLRs: trafficking and regulation. Curr. Opin. Immunol. 44, 26–33 (2017).
De Leo, M. G. et al. Autophagosome-lysosome fusion triggers a lysosomal response mediated by TLR9 and controlled by OCRL. Nat. Cell Biol. 18, 839–850 (2016). This article describes the discovery of a TRLR9–OCRL–TRPML1-mediated mechanism that allows the lysosome to respond to the arrival of autophagic cargo.
Thelen, A. M. & Zoncu, R. Emerging roles for the lysosome in lipid metabolism. Trends Cell Biol. 27, 833–850 (2017).
Ebner, M., Koch, P. A. & Haucke, V. Phosphoinositides in the control of lysosome function and homeostasis. Biochem. Soc. Trans. 47, 1173–1185 (2019).
Folick, A. et al. Ageing. lysosomal signalling molecules regulate longevity in Caenorhabditis elegans. Science 347, 83–86 (2015).
Ramachandran, P. V. et al. Lysosomal signalling promotes longevity by adjusting mitochondrial activity. Dev. Cell 48, 685–696.e5 (2019).
Sardiello, M. et al. A gene network regulating lysosomal biogenesis and function. Science 325, 473–477 (2009).
Settembre, C. et al. TFEB links autophagy to lysosomal biogenesis. Science 332, 1429–1433 (2011). Sardiello et al. (2009) and Settembre et al. (2011) describe the discovery of a lysosomal–autophagic gene network and its master regulator TFEB, the first example of global transcriptional control of lysosomal function.
Hemesath, T. J. et al. Microphthalmia, a critical factor in melanocyte development, defines a discrete transcription factor family. Genes Dev. 8, 2770–2780 (1994).
Palmieri, M. et al. Characterization of the CLEAR network reveals an integrated control of cellular clearance pathways. Hum. Mol. Genet. 20, 3852–3866 (2011).
Willett, R. et al. TFEB regulates lysosomal positioning by modulating TMEM55B expression and JIP4 recruitment to lysosomes. Nat. Commun. 8, 1580 (2017).
Martina, J. A. et al. The nutrient-responsive transcription factor TFE3 promotes autophagy, lysosomal biogenesis, and clearance of cellular debris. Sci. Signal 7, ra9 (2014).
Pastore, N. et al. TFE3 regulates whole-body energy metabolism in cooperation with TFEB. EMBO Mol. Med. 9, 605–621 (2017).
Rega, L. R. et al. Activation of the transcription factor EB rescues lysosomal abnormalities in cystinotic kidney cells. Kidney Int. 89, 862–873 (2016).
Spampanato, C. et al. Transcription factor EB (TFEB) is a new therapeutic target for Pompe disease. EMBO Mol. Med. 5, 691–706 (2013).
Chauhan, S. et al. Pharmaceutical screen identifies novel target processes for activation of autophagy with a broad translational potential. Nat. Commun. 6, 8620 (2015).
Decressac, M. et al. TFEB-mediated autophagy rescues midbrain dopamine neurons from α-synuclein toxicity. Proc. Natl Acad. Sci. USA 110, E1817–E1826 (2013).
Polito, V. A. et al. Selective clearance of aberrant tau proteins and rescue of neurotoxicity by transcription factor EB. EMBO Mol. Med. 6, 1142–1160 (2014).
Xiao, Q. et al. Neuronal-targeted TFEB accelerates lysosomal degradation of APP, reducing Aβ generation and amyloid plaque pathogenesis. J. Neurosci. 35, 12137–12151 (2015).
Pastore, N. et al. Gene transfer of master autophagy regulator TFEB results in clearance of toxic protein and correction of hepatic disease in alpha-1-anti-trypsin deficiency. EMBO Mol. Med. 5, 397–412 (2013).
Settembre, C. et al. TFEB controls cellular lipid metabolism through a starvation-induced autoregulatory loop. Nat. Cell Biol. 15, 647–658 (2013).
Napolitano, G. & Ballabio, A. TFEB at a glance. J. Cell Sci. 129, 2475–2481 (2016).
Martina, J. A., Diab, H. I., Brady, O. A. & Puertollano, R. TFEB and TFE3 are novel components of the integrated stress response. EMBO J. 35, 479–495 (2016).
Nnah, I. C. et al. TFEB-driven endocytosis coordinates MTORC1 signalling and autophagy. Autophagy. 15, 151–164 (2019).
Betschinger, J. et al. Exit from pluripotency is gated by intracellular redistribution of the bHLH transcription factor Tfe3. Cell 153, 335–347 (2013).
Leeman, D. S. et al. Lysosome activation clears aggregates and enhances quiescent neural stem cell activation during ageing. Science 359, 1277–1283 (2018).
Villegas, F. et al. Lysosomal signalling licenses embryonic stem cell differentiation via inactivation of Tfe3. Cell Stem Cell 24, 257–270.e8 (2019).
Curnock, R., Calcagni, A., Ballabio, A. B. & Cullen, P. J. TFEB controls retromer expression in response to nutrient availability. J. Cell Biol. https://doi.org/10.1083/jcb.201903006 (2019).
Pastore, N. et al. Nutrient-sensitive transcription factors TFEB and TFE3 couple autophagy and metabolism to the peripheral clock. EMBO J. 38, e101347 (2019).
Visvikis, O. et al. Innate host defence requires TFEB-mediated transcription of cytoprotective and antimicrobial genes. Immunity 40, 896–909 (2014).
Grey, M. A. et al. Phagocytosis enhances lysosomal and bactericidal properties by activating the transcription factor TFEB. Curr. Biol. 26, 1955–1964 (2016).
Pastore, N. et al. TFEB and TFE3 cooperate in the regulation of the innate immune response in activated macrophages. Autophagy 12, 1240–1258 (2016).
Mansueto, G. et al. Transcription factor eb controls metabolic flexibility during exercise. Cell Metab. 25, 182–196 (2017).
Martina, J. A. & Puertollano, R. Protein phosphatase 2A stimulates activation of TFEB and TFE3 transcription factors in response to oxidative stress. J. Biol. Chem. 293, 12525–12534 (2018).
Nezich, C. L., Wang, C., Fogel, A. I. & Youle, R. J. MiT/TFE transcription factors are activated during mitophagy downstream of parkin and Atg5. J. Cell Biol. 210, 435–450 (2015).
Puertollano, R., Ferguson, S. M., Brugarolas, J. & Ballabio, A. The complex relationship between TFEB transcription factor phosphorylation and subcellular localization. EMBO J. 37, e98804 (2018).
Peña-Llopis, S. et al. Regulation of TFEB and V-ATPases by mTORC1. EMBO J. 30, 3242–3258 (2011).
Martina, J. A., Chen, Y., Gucek, M. & Puertollano, R. MTORC1 functions as a transcriptional regulator of autophagy by preventing nuclear transport of TFEB. Autophagy 8, 903–914 (2012).
Roczniak-Ferguson, A. et al. The transcription factor TFEB links mTORC1 signalling to transcriptional control of lysosome homeostasis. Sci. Signal 5, ra42 (2012).
Settembre, C. et al. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J. 31, 1095–1108 (2012). Peña-Llopis et al. (2001), Martina et al. (2012), Roczniak-Ferguson et al. (2012) and Settembre et al. (2012) demonstrate that the nutrient-regulated mTORC1 kinase complex phosphorylates TFEB and controls its nuclear translocation. This mechanism allows lysosomal function to respond to environmental cues such a nutrient availability.
Vega-Rubin-de-Celis, S., Peña-Llopis, S., Konda, M. & Brugarolas, J. Multistep regulation of TFEB by MTORC1. Autophagy 13, 464–472 (2017).
Di Malta, C. et al. Transcriptional activation of RagD GTPase controls mTORC1 and promotes cancer growth. Science 356, 1188–1192 (2017).
Napolitano, G. et al. mTOR-dependent phosphorylation controls TFEB nuclear export. Nat Commun. 9, 3312 (2018).
Li, L. et al. A TFEB nuclear export signal integrates amino acid supply and glucose availability. Nat. Commun. 9, 2685 (2018).
Silvestrini, M. J. et al. Nuclear export inhibition enhances HLH-30/TFEB activity, autophagy, and lifespan. Cell Rep. 23, 1915–1921 (2018).
Sha, Y., Rao, L., Settembre, C., Ballabio, A. & Eissa, N. T. STUB1 regulates TFEB-induced autophagy-lysosome pathway. EMBO J. 36, 2544–2552 (2017).
Zhang, H. et al. Polyamines control eIF5A hypusination, TFEB translation, and autophagy to reverse B cell senescence. Mol. Cell 30621, S1097–S2765 (2019).
Sakamaki, J. I. et al. Bromodomain protein BRD4 is a transcriptional repressor of autophagy and lysosomal function. Mol. Cell 66, 517–532.e9 (2017).
Kreuzaler, P. A. et al. Stat3 controls lysosomal-mediated cell death in vivo. Nat. Cell Biol. 13, 303–309 (2011).
Martínez-Fábregas, J. et al. Lysosomal protease deficiency or substrate overload induces an oxidative-stress mediated STAT3-dependent pathway of lysosomal homeostasis. Nat Commun. 9, 5343 (2018).
Liu, B. et al. STAT3 associates with vacuolar H+-ATPase and regulates cytosolic and lysosomal pH. Cell Res. 28, 996–1012 (2018).
Shin, H. J. et al. AMPK-SKP2-CARM1 signalling cascade in transcriptional regulation of autophagy. Nature 534, 553–557 (2016).
Chauhan, S. et al. ZKSCAN3 is a master transcriptional repressor of autophagy. Mol. Cell. 50, 16–28 (2013).
Annunziata, I. et al. MYC competes with MiT/TFE in regulating lysosomal biogenesis and autophagy through an epigenetic rheostat. Nat. Commun. 10, 3623 (2019).
Li, Y. et al. Protein kinase C controls lysosome biogenesis independently of mTORC1. Nat. Cell Biol. 18, 1065–1077 (2016).
Garg, S. et al. Lysosomal trafficking, antigen presentation, and microbial killing are controlled by the Arf-like GTPase Arl8b. Immunity 35, 182–193 (2011).
McEwan, D. G. et al. PLEKHM1 regulates autophagosome-lysosome fusion through HOPS complex and LC3/GABARAP proteins. Mol. Cell 57, 39–54 (2015).
Marwaha, R. et al. The Rab7 effector PLEKHM1 binds Arl8b to promote cargo traffic to lysosomes. J. Cell Biol. 216, 1051–1070 (2017).
Antonin, W. et al. A SNARE complex mediating fusion of late endosomes defines conserved properties of SNARE structure and function. EMBO J. 19, 6453–6464 (2000).
Zhao, Y. G. & Zhang, H. Autophagosome maturation: an epic journey from the ER to lysosomes. J. Cell Biol. 218, 757–770 (2019).
Jia, R., Guardia, C. M., Pu, J., Chen, Y. & Bonifacino, J. S. BORC coordinates encounter and fusion of lysosomes with autophagosomes. Autophagy 13, 1648–1663 (2017).
Wang, Z. et al. The Vici syndrome protein EPG5 is a Rab7 effector that determines the fusion specificity of autophagosomes with late endosomes/lysosomes. Mol. Cell 63, 781–795 (2016).
Tabata, K. et al. Rubicon and PLEKHM1 negatively regulate the endocytic/autophagic pathway via a novel Rab7-binding domain. Mol. Biol. Cell 21, 4162–4172 (2010).
Kim, Y. M. et al. mTORC1 phosphorylates UVRAG to negatively regulate autophagosome and endosome maturation. Mol. Cell 57, 207–218 (2015).
Naegeli, K. M. et al. Cell invasion in vivo via rapid exocytosis of a transient lysosome-derived membrane domain. Dev. Cell 43, 403–417.e10 (2017). This study uncovers a mechanism by which lysosome exocytosis helps form an invasive protrusion that breaches tissue barriers during C. elegans development, a process that may be analogous to cancer cell invasion.
Baron, R. et al. Polarized secretion of lysosomal enzymes: co-distribution of cation-independent mannose-6-phosphate receptors and lysosomal enzymes along the osteoclast exocytic pathway. J. Cell Biol. 106, 1863–1872 (1988).
Saffi, G. T. & Botelho, R. J. Lysosome fission: planning for an exit. Trends. Cell Biol. 29, 635–646 (2019).
Levin-Konigsberg, R. et al. Phagolysosome resolution requires contacts with the endoplasmic reticulum and phosphatidylinositol-4-phosphate signalling. Nat. Cell Biol. 21, 1234–1247 (2019).
Krajcovic, M., Krishna, S., Akkari, L., Joyce, J. A. & Overholtzer, M. mTOR regulates phagosome and entotic vacuole fission. Mol. Biol. Cell 24, 3736–3745 (2013).
Wu, H., Carvalho, P. & Voeltz, G. K. Here, there, and everywhere: the importance of ER membrane contact sites. Science 361, eaan5835 (2018).
Friedman, J. R., Dibenedetto, J. R., West, M., Rowland, A. A. & Voeltz, G. K. Endoplasmic reticulum-endosome contact increases as endosomes traffic and mature. Mol. Biol. Cell 24, 1030–1040 (2013).
Dong, R. et al. Endosome-ER contacts control actin nucleation and retromer function through VAP-dependent regulation of PI4P. Cell 166, 408–423 (2016).
Luo, J., Jiang, L., Yang, H. & Song, B. L. Routes and mechanisms of post-endosomal cholesterol trafficking: a story that never ends. Traffic 18, 209–217 (2017).
Wilhelm, L. P. et al. STARD3 mediates endoplasmic reticulum-to-endosome cholesterol transport at membrane contact sites. EMBO J. 36, 1412–1433 (2017).
Kumar, N. et al. VPS13A and VPS13C are lipid transport proteins differentially localized at ER contact sites. J. Cell Biol. 217, 3625–3639 (2018). This article reports that the protein VPS13C, which is mutated in some forms of Parkinson disease, is a lipid transport protein that tethers late endosomes/lysosomes to the ER.
Rocha, N. et al. Cholesterol sensor ORP1L contacts the ER protein VAP to control Rab7-RILP-p150Glued and late endosome positioning. J. Cell Biol. 185, 1209–1225 (2009).
Jongsma, M. L. et al. An ER-associated pathway defines endosomal architecture for controlled cargo transport. Cell 166, 152–166 (2016).
Hong, Z. et al. PtdIns3P controls mTORC1 signalling through lysosomal positioning. J. Cell Biol. 216, 4217–4233 (2017).
Chu, B. B. et al. Cholesterol transport through lysosome-peroxisome membrane contacts. Cell 161, 291–306 (2015).
Starling, G. P. et al. Folliculin directs the formation of a Rab34-RILP complex to control the nutrient-dependent dynamic distribution of lysosomes. EMBO Rep. 17, 823–841 (2016).
Hao, F. et al. Rheb localized on the Golgi membrane activates lysosome-localized mTORC1 at the Golgi-lysosome contact site. J. Cell Sci. 131, jcs208017 (2018).
Wong, Y. C., Ysselstein, D. & Krainc, D. Mitochondria-lysosome contacts regulate mitochondrial fission via RAB7 GTP hydrolysis. Nature 554, 382–386 (2018).
Henne, W. M. et al. Mdm1/Snx13 is a novel ER-endolysosomal interorganelle tethering protein. J. Cell Biol. 210, 541–551 (2015).
John Peter, A. T. et al. Vps13-Mcp1 interact at vacuole-mitochondria interfaces and bypass ER-mitochondria contact sites. J. Cell Biol. 216, 3219–3229 (2017).
González Montoro, A. et al. Vps39 interacts with Tom40 to establish one of two functionally distinct vacuole-mitochondria contact sites. Dev. Cell 45, 621–636.e7 (2018).
Cioni, J. M. et al. Late endosomes act as mRNA translation platforms and sustain mitochondria in axons. Cell 176, 56–72 (2019).
Liao, Y. C. et al. RNA granules hitchhike on lysosomes for long-distance transport, using annexin A11 as a molecular tether. Cell 179, 147–164.e20 (2019). Cioni et al. (2019) and Liao et al. (2019) demonstrate that RNA granules ‘hitchhike’ on late endosomes/lysosomes, enabling them to travel along the axon towards sites of local mRNA translation in the proximity of mitochondria, particularly for synthesis of mitochondrial proteins.
Bonifacino, J. S. & Neefjes, J. Moving and positioning the endolysosomal system. Curr. Opin. Cell Biol. 47, 1–8 (2017).
Encarnação, M. et al. A Rab3a-dependent complex essential for lysosome positioning and plasma membrane repair. J. Cell Biol. 213, 631–640 (2016).
Nakata, T. & Hirokawa, N. Point mutation of adenosine triphosphate-binding motif generated rigor kinesin that selectively blocks anterograde lysosome membrane transport. J. Cell Biol. 131, 1039–1053 (1995).
Harada, A. et al. Golgi vesiculation and lysosome dispersion in cells lacking cytoplasmic dynein. J. Cell Biol. 141, 51–59 (1998).
Burkhardt, J. K., Echeverri, C. J., Nilsson, T. & Vallee, R. B. Overexpression of the dynamitin (p50) subunit of the dynactin complex disrupts dynein-dependent maintenance of membrane organelle distribution. J. Cell Biol. 139, 469–484 (1997).
Matsushita, M., Tanaka, S., Nakamura, N., Inoue, H. & Kanazawa, H. A novel kinesin-like protein, KIF1Bbeta3 is involved in the movement of lysosomes to the cell periphery in non-neuronal cells. Traffic 5, 140–151 (2004).
Guardia, C. M., Farías, G. G., Jia, R., Pu, J. & Bonifacino, J. BORC functions upstream of kinesins 1 and 3 to coordinate regional movement of lysosomes along different microtubule tracks. Cell Rep. 17, 1950–1961 (2016).
Korolchuk, V. I. et al. Lysosomal positioning coordinates cellular nutrient responses. Nat. Cell Biol. 13, 453–460 (2011). A landmark article showing that lysosome positioning regulates mTORC1 signalling, autophagosome formation and lysosome–autophagosome fusion.
Li, X. et al. A molecular mechanism to regulate lysosome motility for lysosome positioning and tubulation. Nat. Cell Biol. 18, 404–417 (2016).
Pu, J. et al. a multiprotein complex that regulates lysosome positioning. Dev. Cell 33, 176–188 (2015).
Filipek, P. A. et al. LAMTOR/Ragulator is a negative regulator of Arl8b- and BORC-dependent late endosomal positioning. J. Cell Biol. 216, 4199–4215 (2017).
Pu, J., Keren-Kaplan, T. & Bonifacino, J. S. A Ragulator-BORC interaction controls lysosome positioning in response to amino acid availability. J. Cell Biol. 216, 4183–4197 (2017). Filipek et al. (2017) and Pu et al. (2017) show that the Ragulator complex engages in a negative regulatory interaction with BORC, mediating perinuclear clustering of lysosomes in response to nutrient deprivation and lysosome dispersal towards the cell periphery in response to epidermal growth factor stimulation.
Raiborg, C. et al. Repeated ER-endosome contacts promote endosome translocation and neurite outgrowth. Nature 520, 234–238 (2015).
Pu, J., Guardia, C. M., Keren-Kaplan, T. & Bonifacino, J. S. Mechanisms and functions of lysosome positioning. J. Cell Sci. 129, 4329–4339 (2016).
Jia, R. & Bonifacino, J. S. Lysosome positioning influences mTORC2 and AKT signalling. Mol. Cell 75, 26–38.e3 (2019).
Clippinger, A. J. & Alwine, J. C. Dynein mediates the localization and activation of mTOR in normal and human cytomegalovirus-infected cells. Genes. Dev. 26, 2015–2026 (2012).
Walton, Z. E. et al. Acid suspends the circadian clock in hypoxia through inhibition of mTOR. Cell 174, 72–87 (2018).
Clippinger, A. J., Maguire, T. G. & Alwine, J. C. Human cytomegalovirus infection maintains mTOR activity and its perinuclear localization during amino acid deprivation. J. Virology 85, 9369–9376 (2011).
Peng, W., Minakaki, G., Nguyen, M. & Krainc, D. Preserving lysosomal function in the ageing brain: insights from neurodegeneration. Neurotherapeutics 16, 611–634 (2019).
Pattison, C. J. & Korolchuk, V. I. Autophagy: ‘self-eating’ your way to longevity. Subcell. Biochem. 90, 25–47 (2018).
Parenti, G., Andria, G. & Ballabio, A. Lysosomal storage diseases: from pathophysiology to therapy. Annu. Rev. Med. 66, 471–486 (2015).
Ballabio, A. & Gieselmann, V. Lysosomal disorders: from storage to cellular damage. Biochim. Biophys. Acta 1793, 684–696 (2009).
Marques, A. R. A. & Saftig, P. Lysosomal storage disorders - challenges, concepts and avenues for therapy: beyond rare diseases. J. Cell Sci. 132, jcs221739 (2019).
Platt, F. M. Emptying the stores: lysosomal diseases and therapeutic strategies. Nat. Rev. Drug Discov. 17, 133–150 (2018).
Platt, F. M., d’Azzo, A., Davidson, B. L., Neufeld, E. F. & Tifft, C. J. Lysosomal storage diseases. Nat. Rev. Dis. Primers 4, 27 (2018).
Dierks, T. et al. Multiple sulfatase deficiency is caused by mutations in the gene encoding the human C(alpha)-formylglycine generating enzyme. Cell 113, 435–444 (2003).
Cosma, M. P. et al. The multiple sulfatase deficiency gene encodes an essential and limiting factor for the activity of sulfatases. Cell 113, 445–456 (2003).
di Ronza, A. et al. CLN8 is an endoplasmic reticulum cargo receptor that regulates lysosome biogenesis. Nat. Cell Biol. 20, 1370–1377 (2018).
Kondo, H. et al. Mutation in VPS33A affects metabolism of glycosaminoglycans: a new type of mucopolysaccharidosis with severe systemic symptoms. Hum. Mol. Genet. 26, 173–183 (2017).
Pavlova E. V. et al. The lysosomal disease caused by mutant VPS33A. Hum. Mol. Genet. 28, 2514–2530 (2019).
van der Beek, J., Jonker, C., van der Welle, R. & Liv, N. Klumperman J. CORVET, CHEVI and HOPS - multisubunit tethers of the endo-lysosomal system in health and disease. J. Cell Sci. 132, jcs189134 (2019).
Settembre, C. et al. A block of autophagy in lysosomal storage disorders. Hum. Mol. Genet. 17, 119–129 (2008).
Lieberman, A. P. et al. Autophagy in lysosomal storage disorders. Autophagy 8, 719–730 (2012).
Seranova, E. et al. Dysregulation of autophagy as a common mechanism in lysosomal storage diseases. Essays Biochem. 61, 733–749 (2017).
Fraldi, A. et al. Lysosomal fusion and SNARE function are impaired by cholesterol accumulation in lysosomal storage disorders. EMBO J. 29, 3607–3620 (2010).
Bartolomeo, R. et al. mTORC1 hyperactivation arrests bone growth in lysosomal storage disorders by suppressing autophagy. J. Clin. Invest. 127, 3717–3729 (2017). This article demonstrates that bone abnormalities in LSDs are caused by mTORC1 activation and consequent autophagy inhibition that suppresses bone growth.
Lim, J. A. et al. Modulation of mTOR signalling as a strategy for the treatment of Pompe disease. EMBO Mol. Med. 9, 353–370 (2017).
Kinghorn, K. J. et al. A Drosophila model of neuronopathic Gaucher disease demonstrates lysosomal-autophagic defects and altered mTOR signalling and Is functionally rescued by rapamycin. J. Neurosci. 36, 11654–11670 (2016).
Maetzel, D. et al. Genetic and chemical correction of cholesterol accumulation and impaired autophagy in hepatic and neural cells derived from Niemann-Pick type C patient-specific iPS cells. Stem Cell Rep. 2, 866–880 (2014).
Fraldi, A., Klein, A. D., Medina, D. L. & Settembre, C. Brain disorders due to lysosomal dysfunction. Annu. Rev. Neurosci. 39, 277–295 (2016).
Nixon, R. A. The role of autophagy in neurodegenerative disease. Nat. Med. 19, 983–997 (2013).
Song, C. Y., Guo, J. F., Liu, Y. & Tang, B. S. Autophagy and its comprehensive impact on ALS. Int. J. Neurosci. 122, 695–703 (2012).
Bras, J. et al. Genetic analysis implicates APOE, SNCA and suggests lysosomal dysfunction in the etiology of dementia with Lewy bodies. Hum. Mol. Genet. 23, 6139–6146 (2014).
Lee, S. M., Chin, L. S. & Li, L. Charcot-Marie-Tooth disease-linked protein SIMPLE functions with the ESCRT machinery in endosomal trafficking. J. Cell Biol. 199, 799–816 (2012).
BasuRay, S., Mukherjee, S., Romero, E. G., Seaman, M. N. & Wandinger-Ness, A. Rab7 mutants associated with Charcot-Marie-Tooth disease cause delayed growth factor receptor transport and altered endosomal and nuclear signalling. J. Biol. Chem. 288, 1135–1149 (2013).
Lee, J. H. et al. Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell 141, 1146–1158 (2010).
Coen, K. et al. Lysosomal calcium homeostasis defects, not proton pump defects, cause endo-lysosomal dysfunction in PSEN-deficient cells. J. Cell Biol. 198, 23–35 (2012).
Lee, J. H. et al. Presenilin 1 maintains lysosomal Ca2+ homeostasis via TRPML1 by regulating vATPase-mediated lysosome acidification. Cell Rep. 12, 1430–1444 (2015).
Shachar, T. et al. Lysosomal storage disorders and Parkinson disease: Gaucher disease and beyond. Mov. Disord. 26, 1593–1604 (2011).
Robak, L. A. et al. Excessive burden of lysosomal storage disorder gene variants in Parkinson disease. Brain 140, 3191–3203 (2017).
Aflaki, E., Westbroek, W. & Sidransky, E. The complicated relationship between Gaucher disease and parkinsonism: insights from a rare disease. Neuron 93, 737–746 (2017).
Spillantini, M. G. et al. Alpha-synuclein in Lewy bodies. Nature 388, 839–840 (1997).
Lee, H. J., Khoshaghideh, F., Patel, S. & Lee, S. J. Clearance of alpha-synuclein oligomeric intermediates via the lysosomal degradation pathway. J. Neurosci. 24, 1888–1896 (2004).
Nixon, R. A. Amyloid precursor protein and endosomal-lysosomal dysfunction in Alzheimer disease: inseparable partners in a multifactorial disease. FASEB J. 31, 2729–2743 (2017).
Wong, Y. C. & Holzbaur, E. L. The regulation of autophagosome dynamics by huntingtin and HAP1 is disrupted by expression of mutant huntingtin, leading to defective cargo degradation. J. Neurosci. 34, 1293–1305 (2014).
Rui, Y. N. et al. Huntingtin functions as a scaffold for selective macroautophagy. Nat. Cell Biol. 17, 262–275 (2015).
Martinez-Vicente, M. et al. Cargo recognition failure is responsible for inefficient autophagy in Huntington disease. Nat. Neurosci. 13, 567–576 (2010).
Freeman, D. et al. Alpha-synuclein induces lysosomal rupture and cathepsin dependent reactive oxygen species following endocytosis. PLOS ONE 8, e62143 (2013).
Ying, J. et al. Lysosomal dysfunction in Down syndrome is APP-dependent and mediated by APP-βCTF (C99). J. Neurosci. 39, 5255–5268 (2019).
Gowrishankar, S. et al. Massive accumulation of luminal protease-deficient axonal lysosomes at Alzheimer disease amyloid plaques. Proc. Natl Acad. Sci. USA 112, E3699–E3708 (2015).
Martini-Stoica, H., Xu, Y., Ballabio, A. & Zheng, H. The autophagy-lysosomal pathway in neurodegeneration: a TFEB perspective. Trends Neurosci. 39, 221–234 (2016).
Boya, P. & Kroemer, G. Lysosomal membrane permeabilization in cell death. Oncogene 27, 6434–6451 (2008).
Appelqvist, H., Wäster, P., Kågedal, K. & Öllinger, K. The lysosome: from waste bag to potential therapeutic target. J. Mol. Cell Biol. 5, 214–226 (2013).
Kimmelman, A. C. & White, E. Autophagy and tumour metabolism. Cell Metab. 25, 1037–1043 (2017).
Perera, R. M. et al. Transcriptional control of autophagy-lysosome function drives pancreatic cancer metabolism. Nature 524, 361–365 (2015).
Perera, R. M., Di Malta, C. & Ballabio, A. MiT/TFE family of transcription factors, lysosomes, and cancer. Annu. Rev. Cancer Biol. 3, (203–222 (2019).
Glunde, K. et al. Extracellular acidification alters lysosomal trafficking in human breast cancer cells. Neoplasia 5, 533–545 (2003).
Bian, B. et al. Cathepsin B promotes colorectal tumorigenesis, cell invasion, and metastasis. Mol. Carcinog. 55, 671–687 (2016).
Monteiro, P. et al. Endosomal WASH and exocyst complexes control exocytosis of MT1-MMP at invadopodia. J. Cell Biol. 203, 1063–1079 (2013).
Dozynkiewicz, M. A. et al. Rab25 and CLIC3 collaborate to promote integrin recycling from late endosomes/lysosomes and drive cancer progression. Dev. Cell 22, 131–145 (2012).
Ylivinkka, I. et al. Motility of glioblastoma cells is driven by netrin-1 induced gain of stemness. J. Exp. Clin. Cancer Res. 36, 9 (2017).
Circu, M. L. et al. A novel high content imaging-based screen identifies the anti-helminthic niclosamide as an inhibitor of lysosome anterograde trafficking and prostate cancer cell invasion. PLOS ONE 11, e0146931 (2016).
Jaishy, B. & Abel, E. D. Lipids, lysosomes, and autophagy. J. Lipid. Res. 57, 1619–1635 (2016).
Mészáros, G., Pasquier, A., Vivot, K., Goginashvili, A. & Ricci, R. Lysosomes in nutrient signalling: a focus on pancreatic β-cells. Diabetes Obes. Metab. 20, 104–115 (2018).
Gilleron, J., Gerdes, J. M. & Zeigerer, A. Metabolic regulation through the endosomal system. Traffic 20, 552–570 (2019).
Gornicka, A. et al. Adipocyte hypertrophy is associated with lysosomal permeability both in vivo and in vitro: role in adipose tissue inflammation. Am. J. Physiol. Endocrinol. Metab. 303, E597–E606 (2012).
Goginashvili, A. et al. Insulin secretory granules control autophagy in pancreatic β cells. Science 347, 878–882 (2015). Starvation of pancreatic β-cells is shown to induce direct fusion of lysosomes with nascent secretory insulin granules by a process that is distinct from conventional autophagy and is controlled by protein kinase D.
Chu, K. Y., O’Reilly, L., Ramm, G. & Biden, T. J. High-fat diet increases autophagic flux in pancreatic beta cells in vivo and ex vivo in mice. Diabetologia 58, 2074–2078 (2015).
Knapp, P. E. & Swanson, J. A. Plasticity of the tubular lysosomal compartment in macrophages. J. Cell Sci. 95, 433–439 (1990).
Lee, S., Sato, Y. & Nixon, R. A. Lysosomal proteolysis inhibition selectively disrupts axonal transport of degradative organelles and causes an Alzheimer’s-like axonal dystrophy. J. Neurosci. 31, 7817–7830 (2011).
Johnson, D. E., Ostrowski, P., Jaumouille, V. & Grinstein, S. The position of lysosomes within the cell determines their luminal pH. J. Cell Biol. 212, 677–692 (2016). This study highlights the heterogeneity of lysosomes by demonstrating that peripherally localized lysosomes have higher pH and lower proteolytic activity than their perinuclearly localized counterparts.
Johnson A. E., Shu H., Hauswirth A. G., Tong A., Davis G. W. VCP-dependent muscle degeneration is linked to defects in a dynamic tubular lysosomal network in vivo. eLife 4, e07366 (2015).
Mrakovic, A., Kay, J. G., Furuya, W., Brumell, J. H. & Botelho, R. J. Rab7 and Arl8 GTPases are necessary for lysosome tubulation in macrophages. Traffic 13, 1667–1679 (2012).
Rubinsztein, D. C., Codogno, P. & Levine, B. Autophagy modulation as a potential therapeutic target for diverse diseases. Nat. Rev. Drug Discov. 11, 709–730 (2012).
Levy, J. M. M., Towers, C. G. & Thorburn, A. Targeting autophagy in cancer. Nat. Rev. Cancer 17, 528–542 (2017).
Yamamoto, S. et al. Autophagy differentially regulates insulin production and insulin sensitivity. Cell Rep. 23, 3286–3299 (2018).
Raiborg C. How nutrients orchestrate lysosome positioning. Contact https://doi.org/10.1177/2515256418756111 (2018).
Ferron, M. et al. A RANKL-PKCβ-TFEB signalling cascade is necessary for lysosomal biogenesis in osteoclasts. Genes Dev. 27, 955–969 (2013).
El-Houjeiri, L. et al. The transcription factors TFEB and TFE3 link the FLCN-AMPK signalling axis to innate immune response and pathogen resistance. Cell Rep. 26, 3613–3628.e6 (2019).
Doronzo, G. et al. TFEB controls vascular development by regulating the proliferation of endothelial cells. EMBO J. 38, e98250 (2019).
Fan, Y. et al. Endothelial TFEB (transcription factor EB) positively regulates postischemic angiogenesis. Circ. Res. 122, 945–957 (2018).
Wada, S. et al. The tumour suppressor FLCN mediates an alternate mTOR pathway to regulate browning of adipose tissue. Genes Dev. 30, 2551–2564 (2016).
Murano, T. et al. Transcription factor TFEB cell-autonomously modulates susceptibility to intestinal epithelial cell injury in vivo. Sci. Rep. 7, 13938 (2017).
Meireles, A. M. et al. The lysosomal transcription factor TFEB represses myelination downstream of the Rag-Ragulator complex. Dev. Cell 47, 319–330.e5 (2018).
Acknowledgements
The authors thank A. De Matteis, G. Diez-Roux, P. Luzio, R. Mattera, G. Napolitano and C. Settembre for comments on the manuscript and J. Goodwin for the original draft of Fig. 3a. Work in A.B.’s laboratory is supported by the US National Institutes of Health (R01-NS078072), the Italian Association for Cancer Research (AIRC) (IG 2018 22103), Fondation Louis-Jeantet and the Telethon Foundation. Work in J.S.B.’s laboratory is supported by the Intramural Program of the Eunice Kennedy Shriver National Institute of Child Health and Human Development, National Institutes of Health (ZIA HD001607).
Author information
Authors and Affiliations
Contributions
The authors contributed equally to all aspects of the article.
Corresponding authors
Ethics declarations
Competing interests
A.B. is a co-founder of CASMA Therapeutics (Boston, MA, USA). J.S.B. declares no competing interest.
Additional information
Peer review informationNature Reviews Molecular Cell Biology thanks R. Puertollano, S. Ferguson, J. Klumperman and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Glossary
- Membrane contact sites
-
Membrane domains where organelles are closely (10–30 nm) held together by tethering proteins.
- RAG GTPases
-
RAGA, RAGB, RAGC and RAGD are small GTPases that belong to a subfamily of the RAS-related GTPases. They act as heterodimers in which RAGA or RAGB interacts with RAGC or RAGD. Their activation by amino acids mediates the recruitment of mechanistic target of rapamycin complex 1 to the lysosomal surface.
- Ragulator
-
Protein complex composed of five subunits (LAMTOR1–LAMTOR5). It is located on the lysosomal surface, where it interacts with RAG GTPases for the recruitment of mechanistic target of rapamycin complex 1 to the lysosome. It also interacts with BORC for the regulation of lysosome positioning.
- Tuberous sclerosis complex
-
(TSC). Protein complex, composed of TSC1 (hamartin), TSC2 (tuberin) and TBC1D7, which functions as a GTPase-activating protein for the small GTPase RHEB.
- Folliculin
-
(FLCN). Tumour-suppressor protein that exists as part of a complex with the folliculin-interacting protein and functions as a GTPase-activating protein for the RAGC and RAGD GTPases.
- Endolysosomes
-
Generic term for various types of endosomes and lysosomes.
- Sphingosine
-
Long-chain unsaturated amino alcohol that forms the backbone of a class of membrane lipids known as sphingolipids.
- Phosphoinositide
-
A class of phospholipids comprising a myo-inositol head group linked by a glycerol moiety to two fatty acyl chains. Phosphoinositides are minor components of cellular membranes involved in signalling and regulation of membrane dynamics.
- 14-3-3 proteins
-
Family of proteins that bind phosphorylated serine or threonine residues on various regulatory proteins such as kinases, phosphatases, transcription factors and signal-transduction proteins.
- Hyperuricaemia
-
Elevated levels of uric acid in blood.
- Lysosomotropic drugs
-
Drugs that concentrate within the acidic lumen of lysosomes and modify lysosomal function.
- Pyroptosis
-
A type of programmed cell death most often triggered by infection with intracellular pathogens and mediated by pore-forming gasdermin D, which permeabilizes the plasma membrane.
- Ferroptosis
-
Iron-dependent form of cell death triggered by inactivation of cellular glutathione-dependent antioxidant defences with consequent accumulation of lipid reactive oxygen species.
- Necroptosis
-
A programmed form of necrosis that is downstream of cell-death receptor signalling, which is generally induced by cell damage but can also be promoted by lysosomal membrane permeabilization.
- Galectin protein family
-
Endogenous carbohydrate-binding proteins (lectins) with specificity for β-galactoside sugars.
- Tripartite motif (TRIM) protein family
-
Family of E3 ubiquitin ligases having RING, B-box and coiled-coil domains.
- AMPK
-
AMP-activated protein kinase that functions as a master regulator of cellular energy metabolism.
- ESCRT machinery
-
Ensemble of complexes known as endosomal sorting complexes required for transport that perform various membrane bending and scission reactions away from the cytoplasm.
- Toll-like receptor (TLR) family
-
Family of transmembrane receptors that recognize pathogen-associated molecular patterns as part of the innate immune response.
- Pathogen-associated molecular patterns
-
Microbial molecules that trigger innate immune responses.
- Nuclear factor-κB
-
Family of transcription factors that regulate the expression of genes involved in both innate and adaptive immunity.
- Interferon regulatory factors
-
Transcription factors that control both innate and adaptive immunity against invading pathogens.
- SNAREs
-
Soluble N-ethylmaleimide-sensitive factor attachment protein receptors, which orchestrate membrane fusion events in the cytoplasm.
- Oleoylethanolamide
-
(OEA). Monounsaturated fatty acid ethanolamide that functions as a bioactive lipid in many physiological processes.
- Nuclear hormone receptors
-
Ligand-activated receptors that bind to specific DNA sequences and regulate gene transcription.
- β-oxidation
-
Catabolic process by which fatty acids are broken down to generate acetyl-CoA in mitochondria.
- MiT-TFE family
-
Family of helix–loop–helix leucine zipper transcription factors that regulate expression of genes involved in the biogenesis and function of lysosomes and autophagosomes.
- Unfolded protein response
-
Cellular stress response triggered by accumulation of unfolded proteins in the endoplasmic reticulum.
- Spermidine
-
Organic polycation that regulates various cellular processes, including translation.
- Vacuolar ATPase
-
(v-ATPase). Multisubunit, ATP-driven proton pump responsible for the acidification of intracellular compartments.
- Tethering complex
-
Protein complex that promotes SNARE-dependent fusion of membrane-bound organelles.
- Atg8/LC3/GABARAP family
-
Homologues of yeast autophagy-related protein 8 (Atg8); ubiquitin-like proteins that function in cargo recruitment to autophagosomes and autophagosome–lysosome fusion.
- RHEB
-
Member of the RAS family of small GTPases that is mainly involved in activation of mechanistic target of rapamycin complex 1.
- Retrograde movement
-
In general, movement from the plus end to the minus end of microtubules; in the axon, movement from the axon terminal towards the cell body.
- Anterograde movement
-
In general, movement from the minus end to the plus end of microtubules; in the axon, movement from the cell body towards the axon terminal.
- CORVET
-
Tethering complex composed of six subunits (VPS3, VPS8, VPS11, VPS16, VPS18 and VPS33) that functions in endosomal fusion events.
- Neurofibrillary tangles
-
Intraneuronal aggregates of hyperphosphorylated tau protein that are most commonly associated with Alzheimer disease.
- α-Synuclein
-
Presynaptic protein pathogenetically linked to Parkinson disease.
- Pompe disease
-
Lysosomal storage disorder caused by acid α-glucosidase deficiency, which results in accumulation of glycogen in the cells.
- Gaucher disease
-
Lysosomal storage disorder caused by deficiency of glucocerebrosidase, which results in the build-up of glucocerebroside in the cells.
- Lewy bodies
-
Abnormal deposits of the protein α-synuclein in the brain of patients with various neurodegenerative disorders, including Lewy body dementia, Parkinson disease and Alzheimer disease.
- Presenilin 1
-
One of the four subunits of the γ-secretase complex. The encoding gene is mutated in an inherited form of Alzheimer disease.
- Amyloid precursor protein
-
(APP). Transmembrane protein that is proteolytically processed to various products, including amyloid-β peptide involved in Alzheimer disease.
- mTORC2
-
Protein complex comprising the mechanistic target of rapamycin kinase and five additional subunits (MLST8, DEPTOR, MSIN1, PROTOR and RICTOR) that functions in the regulation of cellular metabolism and growth.
- Netrin
-
Secreted protein that functions in cell migration and cell–cell and cell–extracellular matrix interactions.
- Anchor cells
-
Cells that participate in the development of the reproductive system in Caenorhabditis elegans.
Rights and permissions
About this article

Cite this article
Ballabio, A., Bonifacino, J.S. Lysosomes as dynamic regulators of cell and organismal homeostasis. Nat Rev Mol Cell Biol 21, 101–118 (2020). https://doi.org/10.1038/s41580-019-0185-4
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41580-019-0185-4
This article is cited by
-
Low-intensity pulsed ultrasound delays the progression of osteoarthritis by regulating the YAP–RIPK1–NF-κB axis and influencing autophagy
Journal of Translational Medicine (2024)
-
A SPLICS reporter reveals \({{{{{\boldsymbol{\alpha }}}}}}\)-synuclein regulation of lysosome-mitochondria contacts which affects TFEB nuclear translocation
Nature Communications (2024)
-
Content-aware frame interpolation (CAFI): deep learning-based temporal super-resolution for fast bioimaging
Nature Methods (2024)
-
Regulation of autophagy by perilysosomal calcium: a new player in β-cell lipotoxicity
Experimental & Molecular Medicine (2024)
-
Advanced oxidation protein products attenuate the autophagy-lysosome pathway in ovarian granulosa cells by modulating the ROS-dependent mTOR-TFEB pathway
Cell Death & Disease (2024)
